Zu Beginn dieser Arbeit standen für inversvoltammetrische Bestimmungen analoge Voltammetriegeräte zur Verfügung, die an Universitäten und anderen Forschungs- und Prüflaboratorien weit verbreitet sind und sicherlich auch zukünftig aufgrund ihrer Robustheit und der einfachen Handhabung eingesetzt werden. Diese Geräte sind für manuelle Anwendungen ausgelegt, verfügen jedoch über TTL-Signaleingänge und sind durch Relaisstellung einer Relais-Karte externen Steuerungen zugänglich. Es besteht somit die Möglichkeit, die Steuerung der inversvoltammetrischen Messung einem Rechner zu übergeben und die Meßwerte mit Hilfe eines A/D-Wandlers zu erfassen.
Können im nächsten Schritt auch alle weiteren, bislang für manuelle Operationen ausgelegten Betriebsmittel für die Inversvoltammetrie an einen Meßwertaufnahmeprozeß adaptiert und aus einem vom System verwalteten Gerätepool zusammengestellt und synchronisiert werden, ist selbst diese komplexe Meßmethode entsprechend Kapitel 6 mit bereits vorhandenen Geräten automatisierbar.
Wird die Ablaufsteuerung zeitgenau realisiert und mit entsprechender Präzision gemessen, ließe sich diese Anwendung auch auf die in Kapitel 6.1 angesprochenen Meßmethoden übertragen und auf weitere Analysenprozesse ausdehnen. Die Wahl der in eine Meßapparatur integrierbaren Geräte ist dann nur durch deren Schnittstelle entsprechend Kapitel 6.3.3 begrenzt.
Die Polarographen “E100“ (BRUKER) und “Polarecord E506“ (METROHM) wurden zunächst mit den Polarographieständen Typ E505 und E554 (METROHM) gekoppelt.
Durch ein Vorschaltgerät (AK-DANNECKER) konnte der zur Anreicherung notwendige Rührmotor und nach Ablauf der Beruhigungsphase das Startsignal (Spannungsvorschub des Polarographen) mittels zweier Schaltuhren in Verbindung mit je einem Relais geschaltet werden.
Die Meßsignale wurden mittels eines XY-Schreibers aufgezeichnet, die Signalhöhen mit einem Lineal ausgemessen und anschließend mit einem Taschenrechner oder PC ausgewertet.
Für beide Geräte wurden Kalomelelektroden mit gesättigter Kaliumchloridlösung als Bezugselektroden, Platindrähte als Gegenelektroden sowie zur inversvoltammetrischen Analyse am hängenden Quecksilbertropfen (HMDE) eine Quecksibertropfenelektrode E410 der Firma METROHM eingesetzt.
Die Größe des Quecksilbertropfens wurde per Hand an einer skalierten Mikrodosierschraube eingestellt.
Folgende Punkte sind bei dieser Gerätekonfiguration zu berücksichtigen:
- Die Polarographiestände enthalten je zwei Gefäße, von denen nur eine Meßzelle je Stand für die Inversvoltammetrie geeignet ist, da hier der Deckel der Zelle in der Mitte für die Quecksibertropfenelektrode (HMDE) zugänglich ist. Auch die Begasung der Meßlösung mit Stickstoff vor der Messung kann ohne Kontaminationsrisiko nur hier erfolgen.
- Die Quecksilbertropfelektrode (HMDE) muß von Hand eingestellt und der Quecksilbertropfen nach erfolgter Messung durch einen leichten Schlag auf das Voltammetriegefäß zum Abfallen gebracht werden. Die Genauigkeit der Tropfenbildung mittels der Mikrodosierschraube hängt daher sehr von der exakten Arbeitsweise des Analytikers ab. Zudem kann der Quecksilberfaden in der Elektrodenkapillare auch durch eine Erschütterung reißen, was sich oft erst während der Messung bemerkbar macht.
- Der Meßbereich beider Polarographen mußte auf eine vorher festgelegte, maximale Amplitude eingestellt werden. Der durch die Zelle fließende Strom konnte nur in den Grenzen des Schreibers (Polarecord E506) oder durch eine begrenzte Spannungsspitze des Analogausganges (BRUKER E100) wiedergegeben werden. Die Simultanbestimmung von sehr unter-schiedlichen Konzentrationen einzelner Metalle in einer Probe war nur durch manuelle Empfindlichkeitsumschaltung möglich. Die Signal-verstärkung der Meßgeräte benötigte jedoch eine gewisse Zeit, um sich auf den gewählten Meßbereich einzustellen und das Signal zu stabilisieren. Vor jeder neuen Probe mußte zudem der in Abhängigkeit der Elektrolytkonzentration und des pH-Wertes sich ändernde Grundstrom auf den Signalausgang abgeglichen werden.
- Die Analysenergebnisse konnten nur durch Eingabe aller Meßwerte in einen Rechner ermittelt werden. Alle eingesetzten Volumina, Verdünnungsschritte und Pufferzugaben mußten in der Berechnung berücksichtigt werden. Bei kleinen Signalen war die Genauigkeit der Meßwerte zudem durch die subjektive Erkennung der Grundlinie limitiert.
- Die manuelle Bedienung der voltammetrischen Meßzelle sowie das exakte Einhalten aller Arbeitsschritte war mit einer hohen Fehlerquote behaftet. Alle Dosierungsschritte, das Einfüllen der Meßlösung, Puffer- und Standardzugaben oder die mehrfache Reinigung der Meßzelle und aller verwendeten Gerätschaften mußte sorgsam von Hand durchgeführt werden.
Die aufgezählten Punkte machen deutlich, daß sich reproduzierbare Meßergebnisse im Spurenbereich kaum erzielen ließen. Bei Verwendung hochreiner Chemikalien und Wasser einer MILLI-Q- Anlage variierten die Blindwerte um ein Vielfaches der nach dem Eichgeradenverfahren errechneten Nachweisgrenzen, die im Optimum für die Elemente Zink, Cadmium, Blei und Kupfer zu 1 µg/l und für Nickel und Cobalt zu 0,5 µg/l ermittelt wurden, jedoch aufgrund der stark schwankenden Blindwerte nicht als repräsentativ angesehen werden konnten.
Die berechneten Nachweisgrenzen können für diesen recht simplen apparativen Aufbau als sehr niedrig angesehen und durchaus dem Spurenbereich zugeordnet werden, sind aber weit höher, als die in der Literatur für die Inversvoltammetrie beschriebenen Nachweisgrenzen (s. Tabelle 4). Dies läßt sich einerseits durch einen ungenauen zeitlichen Ablauf des Meßvorganges (von der Begasung bis zur Messung der letzten Standardaddition) als auch durch die Fehlerquote und die Gefahr einer Kontamination bei manueller Bedienung erklären. Zudem stellte die Wahl des Meßbereiches immer einen Kompromiß zwischen ausreichender Empfindlichkeit für das niedrigste zu erwartende Signal und der größten Amplitude dar, die für ein simultan gemessenes Element durch einen begrenzten Analogausgang oder Schreiber dargestellt werden kann. Die rechnergestützte Weiterverarbeitung der erhaltenen Signale, um z.B. niedrige Signale zu vergrößern, von der Grundlinie zu unterscheiden und präzise auszuwerten, war ebenso wie die Berechnung der Probenkonzentrationen nicht möglich.
Es mußten zunächst nicht nur Vorkehrungen gegen mögliche Kontaminationsquellen getroffen werden (Kapitel 11), sondern der Aufbau des Arbeitsplatzes mußte für eine Routineanalytik und eine schrittweise Automation ausgelegt sein.
Die Messung mit zwei Polarographen an den vorhandenen, nur zur Hälfte nutzbaren Ständen beanspruchte unnötig viel Platz. Die zeitintensive Begasung der Proben mit Stickstoff nahm einen Großteil der Meßzeit in Anspruch (10 min N2-Begasung / 10 ml Probelösung, 6 min inversvoltammetrische Meßzeit) und sollte daher zur Steigerung des Probendurchsatzes extern durchgeführt werden können.
Eine den Messungen vorgeschaltete Probenvorbereitung oder ein Aussäuern von nicht zur Messung eingesetzten Gefäßen mußte ebenfalls an den dafür vorgesehenen Vorrichtungen erfolgen. Um die Kontamination aus der Laborluft zurückzudrängen, sollten die Voltammetriegefäße immer mit den entsprechenden Deckeln dicht verschlossen werden. Es wurde daher ein Stand für die Inversvoltammetrie mit voneinander unabhängigen Meßzellen angefertigt, die alle über eine individuell einstellbare Stickstoffbegasung (Lösung und Überstand) und einen Rührer verfügten. Für jeden Polarographen wurde eine Meßzelle mit den entsprechenden Elektroden ausgestattet.
Die Inversvoltammetrie stellt für die Automation aller Arbeitsschritte besonders hohe Anforderungen an die Steuerung der für die Analyse benötigten Gerätschaften. Aufgrund der Komplexität der Apparatur war es bisher nicht möglich, eine Automation dieses Prinzips zu realisieren. Die Reinigung der Meßzelle stellt dabei ein zentrales Problem dar.
Tab. 9 Ablauf einer Voltammetrischen Analyse |
| Operation | Stellung in der Analyse |
|---|---|
| Spülen der Meßzelle | Analysenvorbereitung |
| Einfüllen der Probe | Analysenvorbereitung |
| Einfüllen eines Puffers | Analysenvorbereitung |
| Begasen der Probe | Analysenvorbereitung |
| Abschlagen eines Quecksilbertropfens | Messung |
| Bildung eines neuen Quecksilbertropfens | Messung |
| Rühren der ProbeĀ | Messung |
| Beruhigen der Probenlösung | Messung |
| Starten des Meßgerätes | Messung |
| Zugabe der Standardlösung | Unterstützung des Analysenverfahrens |
In Tab. 9 werden die einzelnen zu automatisierenden Teilaufgaben aufgeführt. Da die voltammetrischen Bestimmungen im Ultraspurenbereich durchgeführt werden sollen, muß eine Spülung der Meßzelle vor jeder Analyse sorgfältig vorgenommen werden, um eine Verschleppung von Substanzen aus vorherigen Analysen zu verhindern. Das Entfernen der Meßzelle aus der Apparatur zur Reinigung verbietet sich ebenfalls.
Ein geeignetes Gerät zur Durchführung solcher Operationen ist z.B. der Auto-Sampler Modell 222 der Firma Gilson ABIMED, das bisher für einen anderen Einsatz konzipiert war. Dieses Gerät besitzt eine Schlauchpumpe, mit der Flüssigkeiten abgepumpt werden können. Zusätzlich verfügt es über einen Dilutor, mit dessen Hilfe genau dosierte Mengen wäßriger Lösungen zugeführt werden. Die Zuführung geschieht über eine Dosierspitze, die an einem motorgetriebenen Arm befestigt ist, der drei Freiheitsgrade (X,Y und Z) besitzt.
Damit ist es möglich, von einem Probengefäß eine definierte Menge Substanz aufzunehmen und in die Meßzelle einzugeben. Da das Gerät programmierbar ist und über TTL-Eingänge bzw. TTL-Ausgänge verfügt, ist ein Anschluß an einen Rechner über eine Relais-Karte zu realisieren.
Das Begasen der Probe geschieht über ein Gasventil, das mit einem TTL-Signal gesteuert wird.
Um einen nicht mehr benötigten Quecksilbertropfen zu entfernen, wird der Elektrode ein leichter Stoß versetzt und der Tropfen fällt ab. Der Stoß kann durch einen einfachen Magnethammer erzeugt werden, der über ein Relais gesteuert wird.
Zur Bildung eines Quecksilbertropfens drehte bisher der Analytiker eine Mikrodosierschraube an der Arbeitselektrode (HMDE). Diese Mikrodosierschraube wird mit einem Schrittmotor verbunden, der durch eine Steuerkarte mit Hilfe von TTL-Signalen definierte Drehungen durchführt.
Die Zugabe von Standardlösungen wird vom Auto-Sampler durchgeführt.
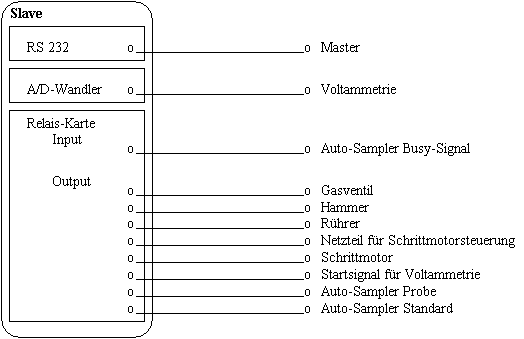 |
Abb.18 Physikalische Verknüpfung der Meßapparatur |
In Abb.18 ist die physikalische Verknüpfung der Meßapparatur dargestellt. Diese Verknüpfung muß dem System bekannt gemacht werden. Hierzu gehört auch die Definition eines Befehlssatzes für jedes einzelne Gerät.
Der Befehlssatz für jedes Gerät besteht aus Elementaroperationen, die in einer externen Datei gespeichert werden und die auf die Anforderungen der einzelnen Meß- und Zubehörgeräte abgestimmt werden können.
Tab.10 Logische Verknüpfung der Meßapparatur |
| Gerät | Befehl | Elementaroperationen |
|---|---|---|
| Auto-Sampler | Probe | Setze TTL-Ausgang 3 high Warte bis TTL-Eingang 13 high Setze TTL-Ausgang 3 low Warte bis TTL-Eingang 13 low |
| Auto-Sampler | Standard | Setze TTL-Ausgang 9 high Warte bis TTL-Eingang 13 high Setze TTL-Ausgang 9 low Warte bis TTL-Eingang 13 low |
| Gasventil | An Aus |
Setze TTL-Ausgang 4 high Setze TTL-Ausgang 4 low |
| Hammer | Abschlagen | Setze TTL-Ausgang 8 high Setze TTL-Ausgang 8 low |
| Netzteil | An Aus |
Setze TTL-Ausgang 6 high Setze TTL-Ausgang 6 low |
| Schrittmotor | 1SKTa | Setze TTL-Ausgang 7 high Setze TTL-Ausgang 7 low Setze TTL-Ausgang 7 high Setze TTL-Ausgang 7 low Setze TTL-Ausgang 7 high Setze TTL-Ausgang 7 low Setze TTL-Ausgang 7 high Setze TTL-Ausgang 7 low Setze TTL-Ausgang 7 high Setze TTL-Ausgang 7 low |
| Rührer | AnĀĀ Aus |
Setze TTL-Ausgang 1 high Setze TTL-Ausgang 1 low |
| Voltammetrie | Messen | Setze TTL-Ausgang 2 high Öffne A/D-Kanal 10 Setze TTL-Ausgang 2 low |
| aSKT = Skalenteil der HMDE-Mikrodosierschraube |
Tab.10 zeigt die implementierten Befehle mit ihren Elementaroperationen für jedes Gerät.
In einer Steuersequenz werden diese Befehle dann verknüpft, um eine zeitlich und funktional exakte Zusammenarbeit aller Gerätschaften zu gewährleisten.
Alle die Messung vorbereitenden Schritte, der Meßvorgang selbst sowie eine Nachbereitung oder Unterstützung des Analysenverfahrens können hier zusammengefaßt werden. Diese Steuersequenz kann dann zusammen mit dem Kalibrierungsmodus (Standardaddition, lineare Kalibrierung oder Kalibrierung mit internem Standard) sowie den Konzentrationseinheiten für die verwendeten Standards und das zu berechnende Ergebnis als Methode abgespeichert werden.
In Tab.11 wird eine typische Steuersequenz für eine vollautomatisierte Voltammetriemessung gegeben. Die dargestellte Steuersequenz beinhaltet über 120 Elementaroperationen und ist trotzdem gut überschaubar.
Tab.11 Steuersequenz |
| Sequenzbefehl | Kommentar |
|---|---|
Gasventil 1 An Rührer 1 An |
Probenvorbereitung |
| Auto-Sampler 1 Probe | Einfüllen einer neuen Probe |
| Timer 300 Gasventil 1 Aus Rührer 1 Aus |
Begasen der Probe |
| NETZ 1 An HMDE 1 1SKT HMDE 1 1SKT HMDE 1 1SKT HMDE 1 1SKT NETZ 1 Aus |
Bildung eines Quecksilbertropfens |
| *********** | Messung |
| HAMMER 1 Abschlagen HAMMER 1 Abschlagen |
Abschlagen des Tropfens |
| NETZ 1 An HMDE 1 1SKT HMDE 1 1SKT NETZ 1 Aus |
Bildung eines Quecksilbertropfens für eine Messung |
| Rührer 1 An Timer 180 Rührer 1 Aus |
Rühren der Lösung über 180 Sekunden |
| Timer 30 | Beruhigen der Lösung über 30 Sekunden |
| Voltammetrie Messen | Messen der Lösung |
| *********** | |
| Rührer 1 An Gasventil 1 An |
Durchmischen der Probe bei Standardzugabe |
| Auto-Sampler 1 Standard | Zugabe der Standardlösung |
| Rührer 1 Aus Gasventil 1 Aus |
Nachdem die Definition und Abarbeitung solcher Sequenzen möglich ist, können alle Befehle in der Steuersequenz oder der Autosampler-programmierung variiert und ohne manuelle Eingriffe unter den gleichen Bedingungen aber mit geänderten Parametern wiederholt werden. Dadurch kann das Meßgerät weitestgehend mit frei wählbarem Zubehör ausgestattet und der Einfluß einzelner Operationen unter reproduzierbaren Bedingungen geprüft und optimiert werden.
Im ersten Schritt wurde die Steuerung der inversvoltammetrischen Messung (Deposition, Beruhigung und Start des Spannungsvorschubes) einem Rechner übergeben und die Meßwerte mit Hilfe eines A/D-Wandlers erfaßt. Die Signalmaxima und Peakgrenzen wurden automatisch gesetzt und die Konzentration der Probenlösung sowie die Erfassungsgrenze berechnet.
Die Stickstoffbegasung, alle Dosierungsschritte, das Einfüllen der Meßlösung, die Tropfenbildung an der Quecksilbertropfelektrode, Puffer- und Standardzugaben sowie die mehrfache Reinigung der Meßzelle wurden von Hand durchgeführt.
Tab.12 Inversvoltammetrische Parameter zur Bestimmung von Zink, Cadmium, Blei und Kupfer |
| Polarographen: | Bruker E 100 und Metrohm E506 |
| Modus:ĀĀĀĀĀĀĀĀĀĀĀĀ | Differential-Puls |
| Quecksilbertropfengröße HMDE: | 2 Skalenteile@1.38mm2 |
| Pulshöhe:Ā | 50 mV |
| Spannungsvorschub: | 10 mV/s |
| Anreicherungszeit (tel ): | 3 min |
| Beruhigungszeit (tRuhe ): | 30 s |
| Anfangspotential: | -1.25 V |
| Endpotential:Ā | 0.25 V |
| Stickstoffbegasung: | 10 min bei 10 ml Lösungsvolumen |
| Empfindlichkeit: | 2,5 µA Vollausschlag |
| Elektrolyt: Ā | 100 µl 4 M Acetatpuffer pH 4.7 ad 10 ml Probelösung 0,1% HNO3 |
Die Korrelationskoeffizienten, Erfassungsgrenzen und die aus der Kalibrierfunktion berechneten Proben- oder Blindwerte konnten jetzt über die Signalhöhe und -fläche ermittelt und miteinander verglichen werden. Es wurden für jede Kalibrierung mindestens 7 Konzentrationen mittels Standardaddition von 1 bis 50 µg/l mit den in Tab.12 aufgeführten Parametern und dem beschriebenen Versuchsaufbau gemessen. Der gebildete Mittelwert mit der Standardabweichung (s) wurde aus 8 Kalibrierversuchen berechnet.
Tab.13 Vergleich der Korrelationskoeffizienten |
|
|
rHöhe |
srHöhe. |
rFläche |
srFläche |
|
Zink |
0.99812 |
0.002682 |
0.99991 |
0.000078 |
|
Cadmium |
0.99805 |
0.001883 |
0.99751 |
0.001494 |
|
Blei |
0.99722 |
0.002097 |
0.99417 |
0.006557 |
|
Kupfer |
0.99836 |
0.001900 |
0.99781 |
0.002837 |
Ein Vergleich der für die Signalhöhe und Signalfläche berechneten Korrelationskoeffizienten (rHöhe und rFläche) bestätigen die theoretischen Grundlagen (s. Tab.13). Bei dem durch das 2. Ficksche Gesetz beschriebenen Stofffluß und dem resultierenden diffusionskontrollierten Strom wird eine peakförmige Kurve erhalten, wobei der maximale Wert des Diffusionsstromes, also die Signalhöhe, der Konzentration der elektrochemisch aktiven Substanz direkt proportional ist.
Nur für das Element Zink wurde ein besserer Korrelationskoeffizient für die Peakfläche ermittelt. In diesem Fall wird das Zink-Signal bei niedrigem pH von einem durch Wasserstoff-Reduktion verursachten Anstieg des Grundstromes überlagert. Das Signal hebt sich mit zunehmender Konzentration immer mehr von diesem Grundstrom ab und bildet eine immer breiter werdende Schulter, bevor die Signalhöhe sichtbar zunimmt.
Trotzdem konnte bei Absicherung des unteren
Arbeitsbereichsendes durch Ermittlung des Prüfwertes Xp (Xp-Höhe
und Xp-Fläche) für alle Elemente eine deutlich niedrigere
Konzentration über die Peakhöhe berechnet werden (s. Tab.14),
wenn auch mit einer großen Standardabweichung (spx).
Tab.14 Vergleich der Prüfwerte Xp (Erfassungsgrenze) [µg/l] |
|
|
Xp-Höhe |
SXp |
Xp-Fläche |
SXp |
| Zink | 0.305 | 2.035 | 3.287 | 2.419 |
| Cadmium | 1.790 | 0.697 | 2.211 | 0.623 |
| Blei | 2.419 | 1.249 | 3.150 | 1.598 |
| Kupfer | 1.563 | 0.896 | 1.810 | 0.712 |
Die über die Kalibrierfunktion berechneten Probengehalte für die Probenlösungen ohne Standardzusatz X0 (Blindwert) ergaben für die Elemente Cadmium, Blei und Kupfer geringere Standardabweichungen sX0 bei Auswertung der Peakhöhen (s. Tab.15).
Tab.15 Vergleich der Blindwerte X0 |
|
|
X0-Höhe |
sX0 |
X0-Fläche |
sX0 |
|
Zink |
0.894 |
0.297 |
0.306 |
0.125 |
|
Cadmium |
0.089 |
0.244 |
0.043 |
0.310 |
|
Blei |
0.571 |
0.531 |
0.656 |
0.756 |
|
Kupfer |
0.148 |
0.467 |
0.228 |
0.577 |
Auch für das Element Zink ist ein Blindwert von 0,9 µg/l, ermittelt über die Peakhöhe, als durchaus realistisch einzuschätzen, zudem besonders offensichtliche Zink- und Bleikontaminationen oftmals zu einem frühzeitigen Abbruch der Analyse führten.
Die in Kapitel 7 ”Realisierung der vollautomatischen analogen Voltammetrie” beschriebene automatisierte Meßapparatur wurde mit den in Tab.12 genannten inversvoltammetrischen Parametern und der in Tab. 11 gegebenen Steuersequenz betrieben.
Die Proben- und Standardzugabe sowie das Spülen der Meßzelle wurde von dem Autosampler durchgeführt. Auf die durch die Steuersequenz ausgelösten TTL-Signale reagierte der Autosampler entsprechend der externen Autosamplersteuerung. Das für jeden Standardschritt zu pipettierende Standardvolumen ebenso wie das Probenvolumen und die Anzahl der Spülzyklen wurde vor Beginn der Analyse in das Autosamplerprogramm eingegeben.
Auch alle anderen zur Messung benötigten Geräte wurden entsprechend der Steuersequenz über die Relaiskarte gesteuert. Die in die Probenliste (s. ID&WEIGHT Anhang I) eingetragenen Proben konnten jetzt vollautomatisch abgearbeitet werden und benötigten keine manuellen Eingriffe mehr.
Um die Leistungsfähigkeit dieses Systems zu untersuchen, wurden zehn salpetersaure Reinstwasserlösungen (0,1% HNO3) mit einem Metallsalzgehalt von 2 µg/l und ohne eine solche Zugabe abwechselnd gemessen. Zu jeder Probelösung wurden hierzu entsprechend dem Standardadditionsverfahren 7 Standardlösungen addiert, wobei jeder Standardschritt 5 mal gemessen wurde. Jede Analyse beinhaltet somit 40 Einzelmessungen und die gesamte Meßreihe bestand aus 400 Einzelmessungen mit insgesamt 1600 Elementsignalen. Die Standardaddition wurde mit progressiver Konzentrationszunahme von 0,5 bis 11,5 µg/l (1, 2, 4, 7.5, 9, 11.5 µg/l) durchgeführt.
Tab.16 Mittelwerte der analogen Meßwerterfassung mit Autosampler |
|
|
X0 |
sxo |
VB |
sVB |
X2ppb |
s2ppb |
|
Zink |
0.795 |
0.407 |
0.377 |
0.077 |
3.637 |
0.307 |
|
Cadmium |
0.012 |
0.073 |
0.354 |
0.056 |
2.166 |
0.074 |
|
Blei |
0.292 |
0.159 |
0.352 |
0.091 |
2.662 |
0.413 |
|
Kupfer |
0.206 |
0.079 |
0.332 |
0.070 |
2.266 |
0.177 |
Für die Elemente Cadmium, Blei und Kupfer konnte mit der vollautomatischen Versuchsdurchführung eindeutig eine Verringerung der Blindwerte X0 (Mittelwert) und der Standardabweichung sX0 der Blindwerte gegenüber der manuellen Versuchsdurchführung erzielt werden (s. Tab.16). Nur für das Element Zink fällt der Vorteil einer automatischen Versuchsdurchführung zunächst nicht so deutlich auf. Dies kann zum einen durch eine latente Kontamination der verwendeten Gerätschaften und Chemikalien erklärt werden, zum anderen aber konnten die Kalibrierversuche mit diesem vollautomatischen Versuchsaufbau bei sichtbaren Verunreinigungen nicht mehr unterbrochen und mit gereinigten Gefäßen nach Austausch der Lösungen wiederholt werden, was bei manueller Versuchsdurchführung in der Regel der Fall war.
Der Mittelwert X2ppb für die Standardlösungen mit einem Gehalt von 2 µg/l je Element zeigt, daß der Elementgehalt für Zink deutlich um den Blindwert erhöht ist und auch für Blei mit einer Kontamination in diesem Konzentrationsbereich gerechnet werden muß. Der Mittelwert des Vertrauensbereiches (VB, bei P = 95%, f=6) und die Standardabweichung des Vertrauensbereiches (sVB) ist für alle Elemente nahezu gleich und als 95%-Prognoseintervall der Analysenergebnisse erfreulich niedrig.
Tab.17 Verfahrensstatistik der analogen Meßwerterfassung mit Autosampler |
|
|
r |
sr |
Xp |
SXp |
Vx0 [%] |
sVxo [%] |
|
Zink |
0.99906 |
0.00040 |
0.558 |
0.1285 |
3.84 |
0.850 |
|
Cadmium |
0.99934 |
0.00017 |
0.480 |
0.0598 |
3.29 |
0.550 |
|
Blei |
0.99829 |
0.00151 |
0.705 |
0.3085 |
4.31 |
1.558 |
|
Kupfer |
0.99933 |
0.00019 |
0.471 |
0.0843 |
3.43 |
0.346 |
Der Verfahrensvariationskoeffizient Vx0 als relatives Präzisionsmaß (mit der Standardabweichung sVx0) ist für alle Elemente ähnlich niedrig, wenn man bedenkt, daß hier alle Fehler einfließen, die zu einer Streuung der Meßwerte um die Regressionsgerade führen können (s. Tab.17). Zu diesen Fehlern zählen z.B. das zeitlich exakte Einhalten des voltammetrischen Meßvorgangs, die Tropfenbildung an der HMDE mittels Schrittmotor, Standardzugaben und Spülvorgänge des Autosamplers sowie eine Drift des Polarographen selbst.
Die Mittelwerte der Korrelationskoeffizienten r (mit der Standardabweichung sr) sowie die deutlich niedrigeren Prüfwerte Xp (mit der Standardabweichung sXp) zur Absicherung des unteren Arbeitsbereichsendes lassen eine Steigerung der Präzision gegenüber der manuellen Versuchsdurchführung gut erkennen.
Die wahlweise Verdünnung einzelner Probelösungen entsprechend der Probenliste (ID & WEIGHT Anhang I) war zu diesem Zeitpunkt noch nicht möglich, da einzelne Probeninformationen aus der vordefinierten Meßreihe mittels TTL-Signalen nicht an den Autosampler übergeben werden konnten. Die Probenvolumina, Standard- und Puffervolumina sowie die Anzahl und das Volumen der Spülschritte mußte dem Autosamplerprogramm vorher bekanntgemacht werden und wurde für jeden Probeneintrag identisch ausgeführt. Ferner war die Autosamplerprogrammierung nicht unerheblich für die Präzision des Analysenverfahrens und bei ungenügender Reinigung der Meßzelle durch den Autosampler auch verantwortlich für einen durch die vorangegangene Messung hervorgerufenen Blindwert (Memoryeffekt).
Um das Kontaminationsrisiko zu minimieren, wurden eine Reihe von Maßnahmen ergriffen und zum größten Teil in die Autosamplerprogrammierung implementiert:
- Das verwendete Milli-Q-Wasser wurde auf 0,1 % mit Salpetersäure angesäuert.
- Alle Lösungen wurden einzeln pipettiert und bei dem Ansaugen in die Autosamplerkapillare durch ein wählbares Luftvolumen von der Verdünnungs- und Spüllösung segmentiert.
- Die teflonbeschichtete Autosamplerkapillare wurde vor jeder Aktion und nach Standardzugaben gespült.
- Um den Memoryeffekt der Meßzelle zurückzudrängen, wurde diese mehrmals mit angesäuertem Milli-Q-Wasser gespült.
- Es wurde mit einem weitaus größeren Volumen Milli-Q-Wasser gespült, als das vorher zur Analyse eingesetzte Probenvolumen. Der Flüssigkeitsstand der Probelösung sollte überschritten und die durch die Stickstoffbegasung verspritzten Tröpfchen der Probelösung sollten entfernt werden können.
- Das Reinigen der Meßzelle wurde in zwei Zyklen unterteilt. Jeder Spülzyklus bestand aus einer wählbaren Anzahl von Spülschritten (Zugabe der Spüllösung über den Dilutor, Pause und Abpumpen der Lösung mit der Minipuls3 Schlauchpumpe). Zwischen den Spülzyklen wurde ein Reinigungsschritt ausgeführt, bei dem ein frei wählbares Volumen einer bereitgestellten zusätzlichen Lösung (meist 1 ml 10-20%ige Salpetersäure) vor Zugabe der Spüllösung (15 ml) in das Voltammetriegefäß pipettiert wurde. Diese Lösung wurde für eine bestimmte Zeit zum Aussäuern im Voltammetriegefäß belassen, bevor der nächste Spülzyklus mit dem nur schwach angesäuerten Milli-Q-Wasser ausgeführt wurde. Ein dauerhafter Betrieb mit hochkonzentrierten Säuren hätte die Zerstörung der Elektroden in der Meßzelle zur Folge gehabt.
- Während der Reinigungsoperationen und zwischen den Messungen (bei Standardzugaben, Wiederholungen) wurden die Stickstoffbegasung und der Rührer angeschaltet.
Die Anzahl der Spülschritte wurde den Probenkonzentrationen bzw. der eingestellten Empfindlichkeit des Meßgerätes angepaßt. Besonders bei Meßreihen mit Proben sehr unterschiedlicher Konzentration konnte auf ein häufiges Spülen mit Aussäuern der Meßzelle nicht verzichtet werden.
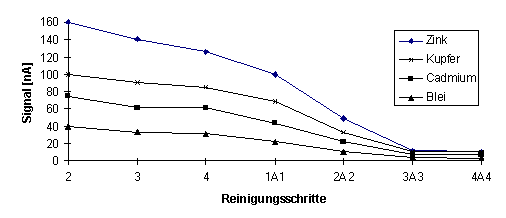 |
Abb.19 Das Signal des Blindwertes ist nach vorhergehender Messung einer 100 µg/l Standardlösung in Abhängigkeit der Spülschritte ohne (2-4) und mit Aussäuern (A) der Meßzelle zwischen den Spülzyklen(1A1-4A4) dargestellt. |
Aus Abb.19 wird deutlich, wie sich das Aussäuern (A) der Meßzelle zwischen den Spülzyklen für den gesamten Reinigungsvorgang auswirkt.
Obwohl bei einem einmaligen Spülen, Aussäuern und wiederholtem Spülen (1A1) ein deutlicher Rückgang der Elementsignale zu verzeichnen ist (hervorgerufen durch die in der Meßzelle verbliebenen Elementspuren der vorherigen Messung), so kann erst ab dreimaligem Spülen vor und nach Aussäuern der Meßzelle (3A3) davon ausgegangen werden, daß der Signalpegel des normalen Blindwertes wieder erreicht wird.
Bei Messungen von Standardlösungen unterschiedlicher Konzentration im Wechsel mit angesäuertem Reinstwasser als Blindwert (Blank) wurde wiederholt ein Memoryeffekt festgestellt (s. Abb.20), obwohl die Konzentration der vorhergehenden Lösung nicht besonders hoch war und die Anzahl der Reinigungsschritte entsprechend Abb.19 hätte ausreichen müssen.
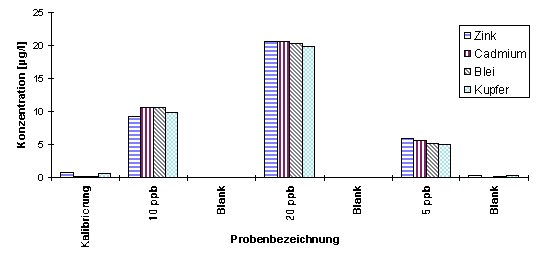 |
Abb.20 Messungen von Standardlösungen im Wechsel mit angesäuertem Reinstwasser |
Die Ursache für das Auftreten solch unkalkulierbar erhöhter Blindwerte sind in erster Linie in der Geometrie der Meßzelle begründet:
- Es wurden 10 ml Probelösung benötigt, damit alle Elektroden in Kontakt zu der Lösung stehen. Da das Voltammetriegefäß für ein maximales Fassungsvermögen von 50 ml ausgelegt ist und auch die Elektroden dieser Größe angepaßt sind, ist die Oberfläche innerhalb der Meßzelle, mit der die Probenlösung in Kontakt kommt, sehr groß und für ein vollständiges Spülen dieser Oberfläche mit dem Autosampler überdimensioniert.
- Der Meßzellendeckel, in dem die Elektroden befestigt sind und der zugleich als Halterung für das Voltammetriegefäß dient, ist für Reinigungs-operationen mit dem Autosampler ohne Öffnen der Meßzelle nicht geeignet und auch für manuelle Reinigungsoperationen, für welche die Meßzelle geöffnet werden muß, nur schwer zugänglich.
Das Reinigen der Meßzelle war somit sehr zeitaufwendig und trotz des hohen Verbrauches an Spüllösung nicht zufriedenstellend. Bei einem Probenvolumen von 10 ml wurden für einen Spülvorgang (3A3 s. Abb.19) bei einer Meßzeit von 6 Minuten (180 sec.Anreicherung, 30 sec. Beruhigung, 150 sec. Spannungsvorschub) die in Tab.18 angegebenen Volumina und Zeiten für eine Analyse je nach Kalibriermethode benötigt:
Tab.18 Zeitbedarf für eine Analyse |
|
Kalibrierung |
Meßzeit |
Spülzeit |
Spülvolumen |
Gesamtzeit |
|
Standardkalibrierung |
Ā 6 Min |
22 Min |
140 ml |
28 Min |
|
Standardaddition |
24 Min |
23 Min |
150 ml |
48 Min |
Blei weist als das elektrochemisch unempfindlichste Element bei der simultanen Messung den höchsten Verfahrensvariationskoeffizienten (Vx0 =4,3 %) und den höchsten Prüfwert (PX = 0,7 µg/l) auf (s. Tab.17). Denn auch wenn die gemessenen Voltammogramme mit dem Auswerteprogramm (s. Anhang I DART bench) nachträglich bearbeitet und selbst kleinste Peaks durch ein Zoom dargestellt werden konnten, so war doch das Auflösungsvermögen des an den 1 V-Analogausgang des Polarographen angepaßten 14 Bit-A/D- Wandlers auf 61 µV begrenzt. Es konnten daher bei einem Meßwert, der einem Zinkgehalt von 22 µg/l entspricht (= 2,5 µA Vollausschlag), simultan keine Bleisignale kleiner 10 ng/l meßtechnisch unterschieden werden. Dies führte bei realen Proben ( z.B. Trinkwasser, Lebensmittel) zu einem Abschneiden der Signale für hohe Zink- und Kupferkonzentrationen (Abb.21b), während die Voltammogramme für die weniger empfindlich meßbaren Elemente Cadmium und Blei, die in diesen Proben meist in wesentlich geringeren Konzentrationen vorliegen, kaum als peakförmige Kurve zu erkennen waren (s. Abb.21a).
| a) | b) | |
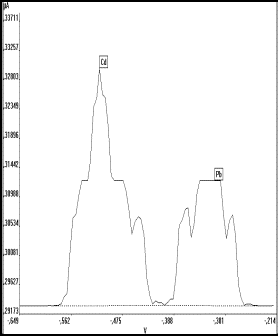 |
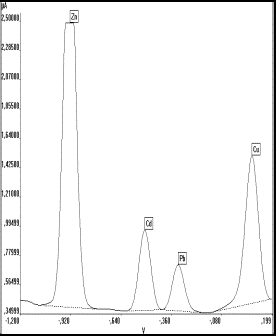 |
|
Abb.21 Voltammogramme von Wasserproben mit a) zu geringer Auflösung b) zu hoher Zn-Konzentration |
||
Die Elementgehalte der untersuchten Probelösungen mußten daher, da bei den verwendeten Polarographen keine automatische Empfindlichkeitswahl (Autoranging) möglich ist, für die simultane Messung in sehr engen Grenzen (s.o.) gehalten werden. Aus der vordefinierten Meßreihe konnten jedoch keine individuellen Verdünnungsschritte an den Autosampler übergeben werden, wodurch die Untersuchung verschiedener biologischer Materialien und Lebensmittel mit sehr unterschiedlichen Elementgehalten im automatischen Betrieb ohne Empfindlichkeitsumschaltung nahezu unmöglich war.