Bei der voltammetrischen Analyse müssen eine Reihe ganz unterschiedlicher Störungen berücksichtigt und ausgeschlossen werden. Bereits in der Probenaufarbeitung können dann bei Kenntnis dieser Störungen Maßnahmen ergriffen werden, die ihnen entgegenwirken oder sie sogar eliminieren.
Auch die Wahl der voltammetrischen Analysenmethode und aller für diese Methode einstellbaren Parameter können bei der Simultanbestimmung von Schwermetallen hinsichtlich der Empfindlichkeit von Bedeutung sein.
Ein prinzipielles Hindernis bei der Steigerung der Empfindlichkeit einer voltammetrischen Analysenmethode stellt das Problem der Unterscheidbarkeit von Faradayschem Strom und Kapazitäts-Strom dar. Um diese beiden Formen von Strom zu unterscheiden, wurden verschiedene instrumentelle Variationen entwickelt. Die differentielle Puls-Voltammetrie erlangte schon früh eine weite Akzeptanz als analytische Methode zur Spurenbestimmung verschiedener Metalle (s. Kapitel 3). Eine weitere Möglichkeit, den Kapazitäts-Strom wirksam zu unterdrücken, bietet die Square Wave Voltammetrie (SWV). Für diese erst wesentlich später durch den kommerziellen Einsatz moderner Mikroprozessoren anwendbare Methode wurden in den 80er Jahren zahlreiche Anwendungen entwickelt, die den Einsatz der Square Wave Voltammetrie als vorteilhaft beschreiben [114].
Für diese Arbeit wurde daher untersucht, welche dieser voltammetrischen Methoden für die vollautomatische Bestimmung von Schwermetallspuren in Lebensmittel- und Umweltproben am besten geeignet ist und mit welchen Parametern und Maßnahmen die Empfindlichkeit gesteigert und evtl. auftretende Störungen beseitigt werden können.
Vor der Durchführung einer polarographischen (voltammetrischen) Analyse ist für die vollständige Beseitigung des störenden Sauerstoffs zu sorgen, der bei Halbstufenpotentialen von 0 bzw. -1V durch Zweielektronenreduktion über Wasserstoffperoxid zu Wasser reduziert wird.
Reaktionen der abgeschiedenen Metalle mit Quecksilber oder mit gleichzeitig abgeschiedenen Depolarisatoren können zur Bildung intermetallischer Verbindungen führen, die je nach ihrer Stabilität die analytische Bestimmung beeinflussen[106]. Im folgenden soll kurz auf die hier relevanten intermetallischen Verbindungen eingegangen werden.
Da Cadmium mit den Edelmetallen Platin, Gold und Silber intermetallische Verbindungen bildet, können die ‘alten’ amalgamierten Pt-, Au- und Ag-Elektroden nur bedingt bei Cadmiumbestimmungen eingesetzt werden. Eine weitere intermetallische Verbindung wird von Cadmium und Kupfer bei Überschreiten der Löslichkeit von Kupfer im Quecksilber gebildet. Es wird angenommen, daß sich das Cadmium unter diesen Bedingungen auf ungelösten Kupferkristallen abscheidet, was zu einer Signaldepression des Cadmiumpeaks bei hohen Cu2+-Konzentrationen führen kann. Diese Cu-Cd Interferenz ist jedoch bei Verwendung der Quecksilbertropfelektrode (HMDE) minimal oder nichtexistent [107].
Störungen bei der Zinkbestimmung durch Bildung intermetallischer Verbindungen von Kupfer und Zink sind ebenfalls in erster Linie bei der Anwendung der Quecksilberfilmelektrode (MFE) beobachtet worden. Hier werden besonders hohe Konzentrationen der Metalle im Quecksilber erreicht, die auch den Einsatzbereich der MFE auf maximal 10 bis 100 µg/l beschränken. Für den hängenden Quecksilbertropfen wird die Bildung der intermetallischen Verbindungen erst in wesentlich höheren Konzentrationen erwartet [14] [108].
Während ARTS et al [109] bei der Verwendung von 0,01 N HNO3 als Grundelektrolyt keine Beeinflussung der Eichkurven beobachten konnten, trat eine deutliche Verringerung der Steilheit für die Zink-Eichkurve bei Anwesenheit von 1500 µg/l Cu in 0,1 N Acetatpuffer auf. Dieses wurde auf die um den Faktor 4 erhöhte Abscheidungseffektivität des Zinks bei Verwendung des Acetatpuffers zurückgeführt, wodurch die Metallkonzentrationen im Quecksilber um diesen Faktor entsprechend höher sind.
In eigenen Untersuchungen konnte dieses Phänomen unabhängig vom verwendeten Elektrotyten bis zu Konzentrationen beider Bindungspartner von 3000 µg/l nicht beobachtet werden. Statt der von ARTS et al [109] eingestellten Tropfengröße (PAR 303, ”small”) wurde jedoch auch ein größerer Tropfen gewählt (PAR 303A, ”medium”), der nach eigenen Erfahrungen konstant und stabil von der Elektrode gebildet wird. Bei bedeutend kleineren Tropfen ist jedoch mit Störungen in höheren Arbeitsbereichen zu rechnen.
Die Wasserstoffreduktion an der Hg-Elektrode kann in saurem Milieu bei der Simultanbestimmung von Zn, Cd, Pb und Cu während der Abscheidung (-1,2V) zur Wasserstoffentwicklung an der Arbeitselektrode und zum Tropfenabfall oder einer Unterbrechung der elektrischen Verbindung des Tropfens zur Quecksilberkapillare führen. Zudem wird das Zinksignal in sauren Lösungen von dem durch die Wasserstoffreduktion verursachten Anstieg des Grundstromes überlagert, wodurch die Auswertung der Signale erschwert oder sogar unmöglich ist. Die Wasserstoffreduktion kann weiterhin durch organische Verunreinigungen oder durch die in Aufschlußlösungen verbliebene Restorganik katalytisch zu positiveren Potentialen verschoben werden, wodurch sich der Verlauf des Grundstromes und die Stabilität des Quecksilbertropfens je nach Matrix und Güte des Aufschlusses ändert. Durch ein Heraufsetzen des Anreicherungspotentials (auf -1,15 V) allein konnte dieser Störung in solchen Fällen nicht begegnet werden.
Stark saure Aufschlußlösungen wurden daher, soweit dies durch die niedrigste zu erwartende Schwermetallkonzentration und die experimentell ermittelten Arbeitsbereichsgrenzen zulässig war, mit Hilfe des Autosamplers verdünnt und durch Zusatz eines Acetatpuffers (300 µl 3,5 M Acetatpuffer, pH 4,7 auf 4 - 5 ml Probelösung) abgepuffert. Obwohl zur Herstellung des Puffers ausschließlich Merck ”Suprapur” Reagenzien verwendet wurden, konnte eine leichte Erhöhung des Blindwertes, besonders für Blei, festgestellt werden. Es wurde daher, besonders bei langen Anreicherungszeiten zur Bestimmung sehr niedriger Schwermetallkonzentrationen, auf die simultane Bestimmung von Zink und die Verwendung eines Puffers verzichtet. Bei späteren Versuchen konnte die zum Aufschluß eingesetzte Salpetersäure mittels eines Eindampfrotors in der Mikrowelle entfernt und der pH-Wert anhand der vorgegebenen Perchlorsäure eingestellt werden, wodurch sich der Einsatz eines Puffers für die Zinkbestimmung erübrigte (s. Kapitel 12.4.2).
Die Bleibestimmung kann durch die Überlagerung von Tl und Sn (II) gestört sein. Bei den üblicherweise zur Probenstabilisierung verwendeten pH-Werten von eins bis zwei ist Sn (II) allerdings nicht stabil und bereits nach wenigen Stunden nicht mehr voltammetrisch nachweisbar. Die Sn(II)-Störung ist somit für die Routineanalytik kein Problem.
Thallium ist oft an der Veränderung des Blei-Signals zu erkennen. Durch Zugabe von EDTA wird Blei komplexiert und Thallium kann dann durch Differenzmessung bestimmt werden [110].
| a) | b) | |
 |
 |
|
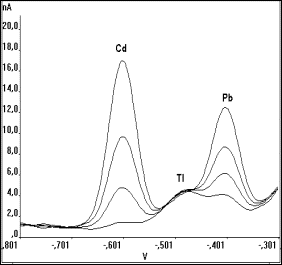
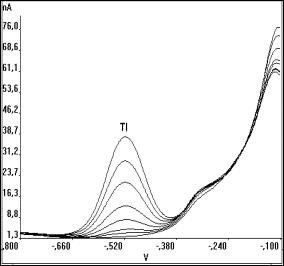
Abb. 24 Thalliumsignale in a) Mineralwasser; neben 0,2 bis 1 µg/l Cd und Pb ohne |
||
In eigenen Untersuchungen konnte meist nach Überprüfung durch Differenz-messungen auf die Bestimmung von Thallium verzichtet werden. Bei der Untersuchung verschiedener Mineralwässer war Thallium eindeutig durch die beschriebene Verformung des Bleisignals zu erkennen (Abb. 24).
Nach Komplexierung des Bleis durch EDTA wurde z.B. ein Thalliumgehalte von 1,1 µg/l in einem Mineralwasser bestimmt, was durch Messungen mit der ICP-MS und GF-AAS nach Anreicherung bestätigt wurde [111]. Die Nachweisgrenze für Tl von 0,2 µg/l (DPASV, tel:300s) ist aber wesentlich höher als für die anderen Elemente (s. Kapitel 10).
Der Arbeitspotentialbereich ist in anodischer Richtung durch die Oxidation des Quecksilbers begrenzt. Das Potential der Oxidation des Elektrodenmaterials wird bei Anwesenheit von Anionen, die mit Hg(I)-Ionen einen schwerlöslichen Niederschlag bilden, in negativer Richtung verschoben [112]. Das Kupfersignal liegt somit besonders in Gegenwart von Chloridionen teilweise weit auf dem Anstieg zur Hg/Hg2Cl2-Flanke. Dies kann zu einer Überschneidung des Elementsignales mit dem durch die Oxidation des Elektrodenquecksilbers verursachten Anstieg des Grundstromes führen, wodurch das Kupfersignal oft gänzlich verdeckt ist oder sich nur geringfügig von dem Grundstrom abhebt. Der Einsatz von Salzsäure zur Probenkonservierung oder zum Aufschluß von Lebensmitteln und anderem Probenmaterial wurde deshalb vermieden.
| (a) | (b) | (c) | ||
 |
||||
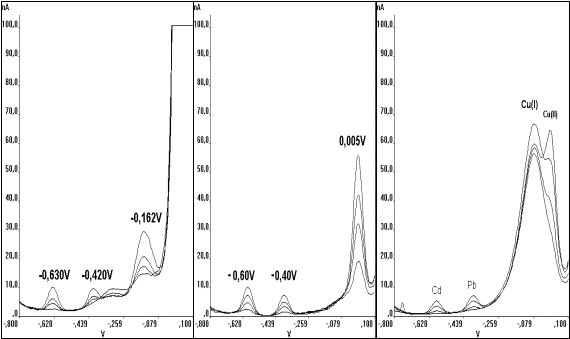
Abb.25 Potentiale innerhalb einer Meßreihe in Abhängigkeit von Salzgehalt und Matrix
a) Meerwasserprobe, b) Standardlösung, c) Gletschereisprobe: |
||||
Das beobachtete Kupfersignal wird z.B. in einer Salzwassermatrix (Abb. 25 a) um -0,177V im Vergleich zu einer Standardlösung (Abb. 25 b) verschoben, während sich die Potentiale von Blei und Cadmium nur geringfügig ändern.
Das in einer geschmolzenen Gletschereisprobe(Abb. 25 c) beobachtete Kupfersignal läßt auf zwei verschiedene Formen von Kupfer schließen, von denen das Signal bei -0,081V einem Kupfer(I)-Komplex und das zweite Signal bei 0,0V dem Kupfer(II) entspricht. Durch UV-Bestrahlung vor der Messung kann bei diesen Proben das organisch komplexierte Kupfer zerstört und anschließend als Kupfer(II) gemessen werden.
Ein weiteres Problem zeigt sich bei der Kupferbestimmung in Aufschlußlösungen, die eine unvollständig zerstörte organische Matrix aufweisen:
| (a) DPASV | (b) DPASV | (C) SWV | ||
 |
 |
 |
||
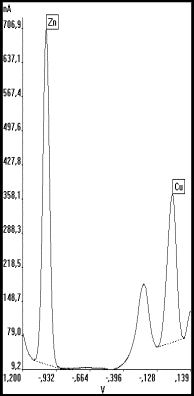
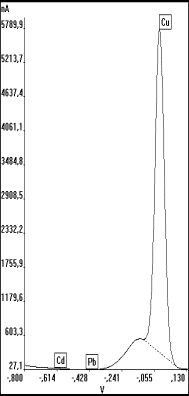
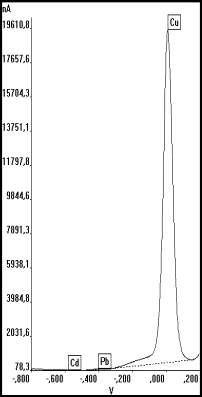
Abb.26 Kupferbestimmung in
Fischleberprobe nach MWE-Aufschluß, |
||||
Für solche biologische Proben und Lebensmittelproben ist grundsätzlich ein vollständiger Aufschluß der organischen Matrix vor der voltammetrischen Messung grundsätzlich erforderlich. Durch die Verwendung von Salpetersäure als hauptsächliches Oxidationsmittel in Druckaufschlußgefäßen aus PTFE wird jedoch nur eine partielle Veraschung erreicht. Durch einen anschließenden UV-Aufschluß oder durch höhere Aufschlußtemperaturen (ca. 300oC) in Quarzgefäßen (HPA s. Kapitel 12.3) kann ein vollständiger Aufschluß für die Voltammetrie erzielt werden. Selbst der Einsatz von Perchlorsäure gewährleistet besonders bei fett- und eiweißhaltigen Proben keinen vollständigen Aufschluß und kann zu Explosionen führen (s. Kapitel 12.2).
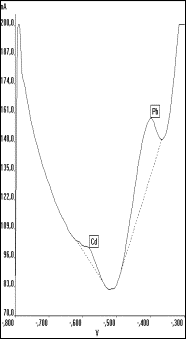
Eine Erhöhung des Grundstromes und dessen Instabilität durch Sauerstoffreduktion und NOx ist in einem Voltammogramm solcher Lösungen sofort zu erkennen und läßt oftmals keine quantitativen Bestimmungen von Cadmium und Blei zu. Selbst Aussagen über vergleichsweise hohe Kupferkonzentrationen können in diesem Fall mit der DPASV nur schwer getroffen werden (s. Abb.26a). Diesem Problem kann durch wesentlich längere Anreicherungszeiten (und entsprechende Verdünnung) bei der DPASV begegnet werden, wodurch die Elementsignale verstärkt und die durch die organischen Bestandteile bedingten Störungen zurücktreten (s. Abb.26b). Besonders vorteilhaft ist in solchen Fällen aber der Einsatz der SWV, da hier die Ströme unterdrückt werden, die langsamen Elektrodenprozessen entsprechen[113]. Durch eine hohe Frequenz (100 Hz) und einen optimalen Spannungsvorschub von 200 mV/s kann der durch Sauerstoff und NOx hervorgerufene Reduktionsstrom nahezu vollständig unterdrückt werden (Abb.26c).
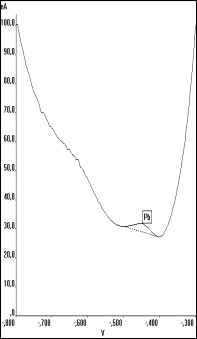
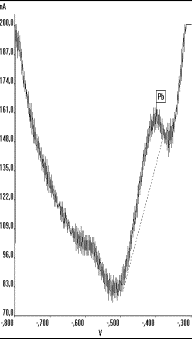
Wie von OSTAPCZUK et al [114] beschrieben, können bei der SWV (Abb.27b) im Vergleich mit der DPASV (Abb.27a) Unregelmäßigkeiten der Basislinie im Potentialbereich des Bleis auftreten, was auf das Alter und den Zustand der Kapillaren zurückgeführt wurde. Die elektrochemischen Eigenschaften der Kapillaren können sich in Abhängigkeit von dem untersuchten Lösungstyp mit der Zeit verschlechtern und dadurch zu einem höheren Untergrund führen. In diesem Fall sei die Bleibestimmung ohne Blindwertkorrektur unpräzise, was jedoch auch durch eine Änderung der Basislinie durch Zusatz der Probenlösung zu analytischen Fehlern führen könne. Es wird daher von OSTAPCZUK et.al [114] nahegelegt, die SWV zur Blei- und Cadmiumbestimmung von geringen Konzentrationen mit neuen Kapillaren und in der Routineanalytik bei höheren Gehalten die Blindwertkorrektur durchzuführen.
Da sich die Grundlinien verschiedener Probelösungen, wie bereits erwähnt, jedoch sehr von der des Blindwertes unterscheiden und sich die elektrochemischen Eigenschaften der Kapillaren innerhalb einer Meßreihe (z.B. über 200 Messungen) bei der vollautomatischen Voltammetrie wesentlich ändern können, ist eine Blindwertkorrektur ebenso wie der Kapillarwechsel innerhalb einer vollautomatisch durchgeführten Meßreihe nicht sinnvoll.
Durch eine mit dem Auswerteprogramm (s. DART bench Anhang I) mögliche Ausschnittsvergrößerung ist zudem zu erkennen, daß die dargestellte ungedämpfte Grundlinie bei der SWV stark durch die Frequenz und die Puls-höhe beeinflußt und besonders bei älteren Kapillaren gestört wird (Abb.27b).
| (a) | (b) | (c) | ||
 |
 |
 |
||
Abb. 27 Ausschnitt der in Abb.26 b) und c) dargestellten Voltammogramme: a): DPASV, b): SWV, c): SWV mit Tiefpaßfilter (Band 5 Hz, Breite 15) |
||||
Durch das Anlegen eines digitalen Tiefpaßfilters niedriger Frequenz können diese höherfrequenten Störungen eliminiert und die Grundlinie soweit geglättet werden, daß eine problemlose Erkennung und Zuordnung der Elementsignale möglich ist (s. Abb.27c).
Sowohl bei der DPASV als auch bei der SWV ist durch Anpassung der Integrationsparameter des Auswerteprogramms bereits eine automatische Auswertung der Meßsignale erreichbar. Sind die Signale wie beschrieben durch hochfrequente Störungen überlagert, so ist durch den Einsatz des digitalen Tiefpaßfilters auch in diesen Fällen eine weitgehend automatische Peakerkennung ausführbar. Bei besonders geringen Elementkonzentrationen oder bei Proben, deren Matrix zu den o.g. Störungen führt, ist eine manuelle Zuordnung der Signalmaxima und Peakgrenzen im Auswerteprogramm möglich und einer Blindwertkorrektur unbedingt vorzuziehen.
[106] Shuman, M.S.; Woodward, G.P.: Intermetallic Compound Formation between Copper and Zinc in Mercury and Its Effects on Anodic Stripping Voltammetry, Anal. Chem. 48 (1976) 1979.
[107] Ostapczuk, P.; Kublik, Z.: J. Elektroanal. Chem. 83 (1977) 1 in [108]
[108] Klahre, P.; Valenta, P.; Nürnberg, H.W.: Ein normiertes pulsinvers-voltammetrisches Analyseverfahren zur Prüfung von Trinkwasser auf toxische Metalle; Vom Wasser 51, (1978) 199.
[109] Arts, W.; Bretschneider, H.J.: Untersuchung der Bleikontamination im Berliner Trinkwasser, deren Ursache sowie Möglichkeiten zur Abhilfe, Dissertation im Fachbereich 21, TU Berlin, (1987).
[110] Gemmer-Colos, V.; Kienast, I.; Trenner, J.; Neeb, R.: Inversvoltammetrische Bestimmung des Thalliums, Fresenius Z. Anal. Chem. 306(1981) 144-149 .
[111] Kerl, W.: Spurenanalytik des Thalliums in Fest- und Flüssigproben mittels GF-Atomabsorptionsspektrometrie, Diplomarbeit am Fachbereich Chemie, AK Danneker, Universität Hamburg (1994).
[112] Henze, G.; Neeb, R.: Elektrochemische Analytik, Berlin-Heidelberg. (1986) S. 88.
[113] Wojciechowski, M.; Go, W.; Osteryoung, J.: Anal.Chem., 57 (1985) 155 in [114].
[114] Ostapczuk, P.; Valenta, P.; Nürnberg, H.W.: Square Wave Voltammetry – A Rapid and Reliable Determination Method of Zn, Cd, Pb, Cu, Ni and Co in Biological and Environmental Samples: J. Elektroanal. Chem., 214 (1986) 51-64.