Bei der Kontrolle der menschlichen Nahrung und anderer biologischer Materialien auf essentielle und toxische Elemente stellt sich das Problem, daß die Mehrzahl der interessierenden Elemente in Konzentrationen zu bestimmen sind, die im Grenzbereich des Nachweisvermögens selbst der leistungs-fähigsten Analysenmethoden liegen.
Auch geringe, nicht mineralisierte Anteile organischer Probenmaterialien oder Artefakte solcher Stoffe führen zu verschiedenartigen Störungen der heute in der Routineanalytik überwiegend angewendeten spektro- und elektro-chemischen Spurenbestimmung. Oft wird eine sinnvolle analytische Aussage dadurch sogar unmöglich gemacht.
Zur Abtrennung der in allen biologischen Materialien im Überschuß vorliegenden organischen Matrix wird dem eigentlichen Elementbestimmungsverfahren in der Regel ein Probenaufschluß vorgeschaltet, der folgende Aufgaben hat [149]:
- Überführung der vielfach undefiniert gebundenen Spurenelemente in definierte lösliche Verbindungen
- Eliminierung der durch die organische Matrix verursachten Analysenstörungen, die oft eine Quelle systematischer Fehler sind
- Isoformierung von Proben- und Kalibriermaterialien
Die Voltammetrie als substanzspezifische Analysenmethode stellt bei der Analyse von biologischem Probenmaterial besonders hohe Anforderungen an das Probenvorbereitungsverfahren. Die Spurenmetalle in der Meßlösung müssen in definierter Form vorliegen und einen weitgehend reversiblen Elektrodenprozeß der Metalle gewährleisten.
Zudem sollte die Lösung frei von Substanzen sein, welche den invers-voltammetrischen Bestimmungsvorgang nachteilig beeinflussen können [130] wie:
- Sauerstoff
- Wasserstoffperoxid
- Komplexbildner
- Tenside und andere auf die Elektrode aufziehende Substanzen
- Viskositätserhöhende Stoffe.
Für die Analyse biologischer Probenmaterialien ist daher das Überführen schwerlöslicher Substanzen in säure- oder wasserlösliche ionogene Verbindungen und einer chemischen Veränderung der Ausgangssubstanz bzw. der Matrix insgesamt zwingend erforderlich. Dies ist bei dieser Probenart nur mit den sehr drastischen Probenvorbereitungsverfahren möglich, auf deren Vor- und Nachteile in den folgenden Kapiteln über Druckaufschlüsse besonders eingegangen werden soll.
Bei der voltammetrischen Schwermetallanalytik von Wasser, Abwasser oder auch flüssigen Lebensmitteln wie Wein und Bier genügen oftmals weniger drastische Maßnahmen, um z.B. die in den verschiedenen Wassertypen enthaltenen Stoffe sowie gelöstes organisches Material (Dissolved Organic Material, DOM) zu zerstören. Hier wird der oxidative UV-Aufschluß eingesetzt, bei dem nur geringe Säuremengen neben Wasserstoffperoxid benötigt werden und der ein zentrales Probenvorbereitungsverfahren in der aquatischen Spurenanalyse darstellt. Die UV-Strahlung kann, wie in dieser Arbeit, u. a. auch für einen ergänzenden Aufschluß bereits teilaufgeschlossener Feststoffproben genutzt werden, weshalb auf die Entwicklung und Anwendung einer UV-Aufschlußapparatur in einem eigenen Kapitel eingegangen wird.
Biologische Probenmaterialien werden vorzugsweise mit hohen Konzentrationen oxidierender Säuren umgesetzt, wobei der Kohlenstoff der organischen Matrix zu Kohlendioxid oxidiert und so von den interessierenden Elementspuren abgetrennt wird. Die wichtigsten Aufschlußreagenzien sind Salpetersäure und Perchlorsäure. Sie werden entweder allein, im Gemisch miteinander oder in Mischungen mit Schwefelsäure eingesetzt.
Von diesen Reagenzien ist Perchlorsäure das stärkste Oxidationsmittel für organisches Material. Das Arbeiten mit Perchlorsäure bringt jedoch auch einige Nachteile mit sich. Ihre hohe Reaktivität gegenüber organischem Material bewirkt zwar eine gute Aufschlußleistung, eine direkte Anwendung muß jedoch unterbleiben, da Perchlorsäure mit organischen Materialien auch explosionsartig reagieren kann. Daher darf sie nur in Gemischen mit anderen Säuren, vor allem mit Salpetersäure, eingesetzt werden. Aber auch dann ist das Arbeiten nicht ungefährlich [131].
Wesentlich sicherer ist das Arbeiten mit Salpetersäure. Allerdings reicht ihr Oxidationsvermögen nicht aus, um biologische Probenmaterialien im offenen System vollständig zu mineralisieren. Je nach Probenmaterial bleiben zwischen 2 und 50% des ursprünglich vorhandenen Kohlenstoffs im Aufschlußrückstand zurück, wobei der Kohlenstoffumsatz teilweise unreproduzierbar ist [132].
Da die Reaktivität der Salpetersäure geringer ist als die der Perchlorsäure, tritt eine explosionsartige Reaktion bei dem Kontakt mit organischem Material nicht auf. Salpetersäure läßt sich in geschlossenen Systemen einsetzen. Zudem kann die handelsübliche analysenreine Säure leicht durch isotherme Destillation nachgereinigt werden [118].
Wie bereits einleitend erwähnt, stellt der oxidative UV-Aufschluß in der aquatischen Spurenanalyse ein zentrales Probenvorbereitungsverfahren für die Voltammetrie dar. Gemäß dem Deutschen Einheitsverfahren zur Wasser-, Abwasser- und Schlammuntersuchung DIN 38406 E 16 [133], wird die Vorbehandlung zur voltammetrischen Bestimmung des Gehaltes an gelöstem Metall mittels UV-Bestrahlung durchgeführt.
Der Analysengang muß bei den verschiedenen Techniken der differentiellen Pulsinversvoltammetrie zur Vermeidung systematischer Fehler (Kontamination und Verluste) schon während der vorausgehenden Probenahme und Probenvorbereitung auf das untersuchte Wasser abgestimmt sein. Je nach Typ des untersuchten Wassers sind nach der Probenahme mehr oder weniger umfangreiche Probenvorbereitungsschritte erforderlich (s. Abb. 43).
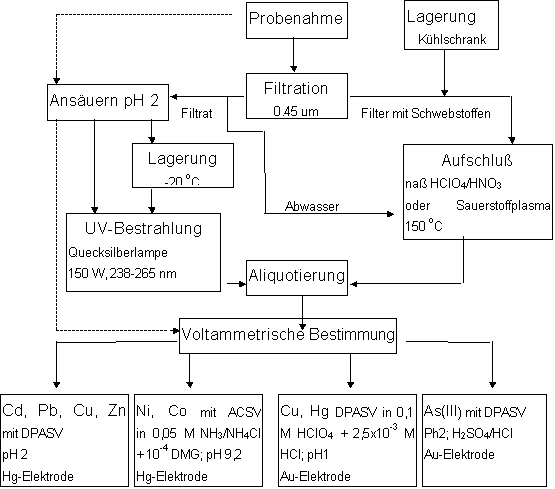 |
Abb.43 Fließchema Metallspurenanalyse verschiedener Wassertypen |
Die Abtrennung meßbarer Schwebstoffgehalte erfolgt durch Filtration. In der aquatischen Spurenchemie wird gemäß der üblichen Konvention ein Membranfilter von 0,45 µm Porenweite eingesetzt [134]. Bei Trinkwasser und Wasser aus den Ozeanen sind in der Regel die Schwebstoffgehalte so gering, daß die Filtration auch im Hinblick auf ein gewisses Kontaminationsrisiko entfallen kann.
Anschließend wird das Filtrat auf pH 2 angesäuert, wodurch die biologische Aktivität noch vorhandener Bakterien unterbunden und die Gewässerprobe wesentlich haltbarer ist, vor allem wenn sie bis zur Analyse im Dunkeln bei - 20 oC gelagert wird.
Sollen Untersuchungen über die chemischen Bindungsformen (speciation) [135] [136] [137] durchgeführt werden, so muß natürlich jegliche pH-Änderung im Filtrat unterbleiben. Die Proben müssen dann möglichst rasch der Untersuchung zugeführt werden, da sie nach kürzeren Zeiträumen eine Veränderung der Speciesverteilung erleiden.
Trinkwasser, meist auch Quellwasser und Wasser aus klaren
Bächen und einer Reihe von Bereichen der Ozeane sowie häufig Regen und Schnee
enthalten vernachlässigbare Konzentrationen an gelöster organischer Materie. In
Wasser aus Flüssen, Seen und Küstenzonen, zuweilen auch in mit organischen
Substanzen verunreinigtem Regen und Schnee, ist das nicht der Fall. Da
Komponenten der gelösten organischen Materie Schwermetalle in stabilen
Komplexen binden, die der voltammetrischen Bestimmung schwer oder gar nicht
zugänglich sind, muß die gelöste organische Materie zerstört werden. Das ist
durch Photolyse möglich, indem man das Filtrat für eine Stunde oder mehr einer
UV-Bestrahlung unterwirft [138]
[139]
[140].
Der Zusatz von etwas
konzentriertem H2O2 beschleunigt die photolytische
Zersetzung der gelösten organischen Materie. Auch die nicht unerheblichen
Mengen organischer Materie in Abwässern können so zerstört werden. Für diesen
Wassertyp bietet sich aber auch der Naßaufschluß mit HClO4/HNO3
an, der auch für Filterproben mit den abfiltrierten Schwebstoffen eingesetzt
werden kann [133].
Der Nachteil des
Naßaufschlusses ist jedoch der hohe Zeit- und Reagenzienverbrauch. Bei reinen,
natürlichen Wässern führt der Zusatz der benötigten Säuremenge zudem leicht zu
Kontaminationsproblemen, was sich durch einen Anstieg der Blindwerte bemerkbar
macht.
Im Unterschied zu den naßchemisch-thermischen Aufschlußverfahren werden bei UV-Aufschlüssen nur geringe Säuremengen neben Wasserstoffperoxid benötigt, wodurch die Kontamination infolge von Schwermetallgehalten aus den Reagenzien gering gehalten werden kann. Durch die UV-Strahlung entstehen sehr reaktive chemische Radikale und es wird Ozon erzeugt.
In Gegenwart von Nitrat wird das voltammetrisch aktive Nitrit während der UV-Bestrahlung gebildet. Dieses stört die Bestimmung und führt zu einem breiten Signal, dessen Anstieg sich mit dem Zn-Peak überschneidet. Nitrit kann zu molekularem Stickstoff reduziert werden, indem nach dem Aufschluß Amido-schwefelsäure (25 µl je 10 ml Probe einer Lösung von 40 g/l H2NSO3H) [143] oder Hydroxylammoniumsulfat ((HONH3)2SO4) [142] in die noch heiße Lösung (80 oC) pipettiert wird.
Quecksilber-Niederdrucklampen verfügen über ein Linienspektrum mit einem hohen Anteil an harter UV-Strahlung bei 185 nm (8% relative Intensität), während die Hauptkomponente ihrer Strahlung entsprechend der Resonanzlinie des Quecksilbers bei 254 nm (90% relative Intensität) liegt. Aufgrund ihrer geringen Wärmeentwicklung können die Quecksilber-Niederdrucklampen als Tauchlampen verwendet werden, weshalb jedoch immer nur eine Probe aufgeschlossen werden kann. Zudem steigt die Kontaminationsgefahr durch das Eintauchverfahren in der Spurenanalytik. Da gelöstes organisches Material (DOM) indirekt durch OH-Radikale und nicht direkt durch die UV-Strahlung abgebaut wird [130], spielt die Strahlungskomponente der Niederdrucklampen bei 185 nm zum Aufschluß organischer Verbindungen keine wichtige Rolle.
Mit Quecksilber-Hochdrucklampen kann hingegen ein Breitbandspektrum von ca. 200 nm bis 436 nm beobachtet werden. Dieses resultiert aus den typischen Quecksilberlinien bei 254 nm, 313nm und 366 nm und geht einher mit einer kraftvollen, kontinuierlichen Wärmestrahlung. Zudem haben Hochdrucklampen eine wesentlich höhere Intensität (Faktor 103) und einen größeren Strahlungsfluß [143] bei allen Wellenlängen (Faktor 10 - 103).
Die Schwierigkeit bei der Nutzung der Quecksilber-Hochdrucklampen ist ihre hohe Hitzeentwicklung, die zu einem hohen Verdampfungsverlust der Probe führen kann und durch eine entsprechende Kühlung kompensiert werden muß.
Kann das Problem der entsprechenden Kühlung bei der Arbeit mit Quecksilber-Hochdruckstrahlern gelöst werden, kommen folgende Vorteile im Vergleich zur Arbeit mit Niederdruckstrahlern zum Tragen:
- höhere Intensität und größerer Strahlungsfluß
- geringere Kontamination durch kontaktlose Bestrahlung
- gleichzeitiger Aufschluß mehrerer Proben
- die Temperatur kann erhöht werden.
Auch in dem DIN-Verfahren 38406 – E 16 wird ein Hg-Hochdruckstrahler mit einer Leistungsaufnahme von mindestens 500 W und einem durchschnittlichen Strahlungsfluß von mindestens 150 W zwischen Wellenlängen von 200 nm und 600 nm gefordert.
Die Proben sind vor der Bestrahlung mit HCl oder HNO3 auf einen pH von 1,7 – 2,0 anzusäuern und werden in halbstündigen Abständen während der Bestrahlung mit 50 µl H2O2 je 25 ml Filtrat versetzt. Je nach DOM-Belastung wird bis zu 3 h bestrahlt, wobei die Temperatur auf mindestens 65 oC ansteigen soll.
KOLB et al [143] nutzten einen Hg-Hochdruckstrahler mit 500 W (705 UV Digestor METROHM) zur gleichzeitigen Bestrahlung von 12 Proben, der sowohl mit einer Wasser- als auch mit einer Luftkühlung ausgestattet ist. Bei dem Aufschluß von Modellwässern konnte festgestellt werden, daß ein Temperaturanstieg auf 65 oC zur Bestimmung von Ni und Co nicht ausreicht. Erst oberhalb Temperaturen von 90 oC kann eine ausreichend hohe Konzentration von OH-Radikalen erwartet werden, mit der ein Aufschluß zur nachfolgenden ACSV-Bestimmung dieser Metalle erfolgreich ist. Selbst stark mit DOM beladene Wässer können so nach KOLB et al [143] innerhalb einer Stunde sicher aufgeschlossen werden.
Bei galvanischen Bädern oder Kokereiabwässern stößt die UV-Photolyse jedoch auch mit wesentlich längerer Bestrahlungsdauer an ihre Grenzen.
In dieser Arbeit wurde der UV-Aufschluß hauptsächlich für die Beseitigung der Restorganik in biologischen Probenmaterialien nach vorangegangenem Druckaufschluß konstruiert und eingesetzt.
Da die für die Voltammetrie kommerziell erhältlichen UV-Aufschlußapparaturen verhältnismäßig teuer sind (> 16000 DM), sollte ein im Handel erhältlicher Quecksilber-Hochdruckstrahler verwendet werden.
Dieser Quecksilber-Hochdruckstrahler muß den beschriebenen Leistungs-anforderungen entsprechen, zentral in eine Probenhalterung zur gleichzeitigen Bestrahlung mehrerer Proben eingebaut werden und zudem mit einem entsprechenden Netzgerät kombinierbar sein.
Für die Konstruktion der UV-Aufschlußapparatur wurde daher der Quecksilber-Hochdruckstrahler TQ 718 (Heraeus Nobelight GMBH, Kleinostheim) ausgewählt, der über das charakteristische Hg-Linienspektrum und eine Leistungsaufnahme von 700 W verfügt (s. Tab. 32).
Tab.32 Strahlungsfluß q des Hg-Hochdruckstrahlers TQ 718 bei 700 W nach[144] |
|
l (nm) |
Strahlungsfluß q (W) |
l (nm) |
Strahlungsfluß q (W) |
|
238 |
4.8 |
313 |
20.1 |
|
248 |
3.2 |
334 |
2.4 |
|
254 |
18.6 |
366 |
30 |
|
265 |
6.6 |
390 |
0.3 |
|
270 |
2.6 |
405 |
14.9 |
|
275 |
1.2 |
436 |
19.7 |
|
280 |
3.2 |
492 |
0.3 |
|
289 |
2.1 |
546 |
23.9 |
|
297 |
4.7 |
577 |
22.1 |
|
302 |
8.6 |
|
|
Der Strahlungsfluß q
ist zwischen 200 nm – 600 nm bei einer |
Durch Zusätze an Metallhalogenid (Z1 – Z4) können die Strahlungs-schwerpunkte im Spektrum verändert werden. Die Gesamtstrahlungsausbeute der Strahler wird dadurch jedoch nicht erhöht. Im Vergleich zum reinen Hg-Hochdruckstrahler werden durch die Metallhalogenidzusätze lediglich die Spektrallinien des Quecksilbers vorzugsweise im kurzwelligen Bereich in ihrer Intensität vermindert [144].
Der Strahler wurde in einem Stahlzylinder (Durchmesser = 22 cm) von einem Keramikträger gehalten, an dem auch die Halterungen für die Probenröhrchen befestigt sind (Abb. 44).
Als Probenröhrchen wurden Quarzgläser verwendet, die mit einem Stopfen und zusätzlich mit einem Deckel gegen die Umluft abgeschirmt sind. Ein Druckausgleich war durch eine Rinne in Stopfen und Deckel möglich.
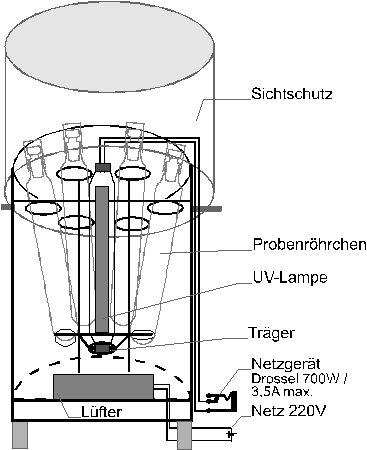 |
Abb.44 UV-Aufschlußapparatur mit 6 Probenröhrchen |
Die Kühlung der Proben konnte durch die Montage zweier Lüfter (z.B. Papst, St. Georgen) übereinander gewährleistet werden. Strahler und Probenröhrchen wurden somit ausschließlich von dem durch die Lüfter eingebrachten Luftstrohm gekühlt. Bei einer Temperatur von 86 ± 2oC konnte ein Verdampfungsverlust innerhalb von 2 Stunden nahezu ausgeschlossen werden. Für diesen Fall wurden die Probenröhrchen markiert und konnten nach längerer Bestrahlungsdauer mit Reinstwasser wieder auf das ursprüngliche Volumen gebracht werden.
Zum Schutz vor UV-Strahlung wurde der Aufschlußapparatur eine lichtundurch-lässige Röhre aufgesetzt, die den Luftstrom nicht beeinträchtigt und nach oben hin offen ist.
Als Netzgerät konnte eine 700 W Drossel in Verbindung mit einem Ringtrafo genutzt werden, bei dem die Spannung zum Starten der Lampe kurzzeitig erhöht wurde.
Um die Anwendung der Salpetersäure auch oberhalb ihres Siedepunktes zu ermöglichen, wurden Druckaufschlußsysteme entwickelt, bei denen die Probe zusammen mit konzentrierter Säure im druckdichten System einem Temperaturprogramm unterworfen wird.
Die Mehrzahl der heute erhältlichen Systeme leitet sich von den u.a. von TÖLG et al[145] beschriebenen Apparaturen ab. Die „Aufschlußbombe“ besteht aus einem PTFE-Gefäß, das in einen Edelstahlzylinder eingesetzt ist und durch einen auf das Gefäß gepreßten PTFE-Deckel abgedichtet wird. Um sicherzustellen, daß es nicht infolge eines zu starken Druckanstiegs zu einem Bersten kommen kann, können die Reaktionsgase bei zu hohem Druck den Deckel gegen einen Federdruck oder eine Berstscheibe öffnen und somit entweichen. Beheizt wird die Bombe durch einen Metallheizblock oder Luftthermostaten[146].
Das Arbeiten in einem geschlossenen System bringt gegenüber dem Aufschluß im offenen System Vorteile:
- Keine Elementverflüchtigungen
- Verkürzung der Aufschlußzeit und Verbesserung des Aufschlusses durch Anwendung von Temperaturen oberhalb des Siedepunktes des Aufschluß-reagenzes
- Geringerer Reagenzienbedarf und dadurch bedingte geringere Reagenzienblindwerte
- Keine Kontamination von außen.
Die Aufschlußtemperatur wird jedoch durch das Gefäßmaterial begrenzt: PTFE läßt, je nach Qualität, Temperaturen von 160 bis 200 oC zu [147]. Bei höheren Temperaturen beginnt es zu erweichen. Auch diffundieren einige Elemente bei hohen Temperaturen und Druck in die Gefäßwandungen hinein, was Memoryeffekte und somit systematische Fehler zur Folge haben kann [148].
Da eine Temperaturerhöhung von 180 auf 200 oC zu keiner nennenswerten Verbesserung des Aufschlusses bezüglich des umgesetzten Kohlenstoffanteils führt und PTFE nicht höher erhitzt werden kann, ist eine Temperatur von 180 oC für den Aufschluß in PTFE-Gefäßen optimal und stellt eine obere Grenze dar[132].
Unabhängig von der Art des aufzuschließenden Materials (Tab. 33) reichen 2 ml HNO3 conz. je 100 mg reinen Kohlenstoffanteils in jedem Falle aus, wenn bei 170 bis 180 oC über 3 Stunden aufgeschlossen wird [132] [149].
Neben den Parametern Temperatur, Zeit und Säuremenge ist jedoch auch das Verhältnis von Probeneinwaage zu Gefäßvolumen für einen möglichst vollständigen Umsatz organischer Probenbestandteile wichtig. Da das durch Reduktion der Salpetersäure freigesetzte NO2 an den Abbaureaktionen organischer Probenbestandteile maßgeblich beteiligt ist, folgt für die praktische Anwendung des Druckaufschlusses, daß man ein Verhältnis von Probeneinwaage zu Tiegelvolumen von 1,4 mg C/ml nicht unterschreiten sollte [149].
Unter diesen Bedingungen lassen sich biologische Materialien zu klaren Lösungen aufschließen. In manchen Proben enthaltene Silikate (z.B. Pflanzen und Muscheln) können jedoch nur mit einer Mischung aus Salpeter- und Flußsäure in Lösung gebracht werden. Die nach einem solchen Aufschluß häufig vorliegenden unlöslichen Verbindungen (CaF2, Fluorosilikate) können in lösliche Nitrate überführt werden, indem im Anschluß an den Druckaufschluß zur Trockne eingeengt und in einigen Fällen noch etwas HNO3 zugesetzt wird [149], wodurch jedoch die Gefahr einer möglichen Kontamination gegeben ist.
Tab33 Kohlenstoffgehalte gefriergetrockneter biologischer Probenmaterialien [149]
|
Probenmaterial |
Kohlenstoffgehalt (%) |
|
Pflanzliche Stoffe |
|
|
Braunalgen |
35 |
|
Weizenmehl |
45 |
|
Spinat |
38 |
|
Pappelblätter |
44 |
|
Buchenblätter |
48 |
|
Ahornblätter |
48 |
|
Gras |
39 |
|
Klee |
36 |
|
Kiefernnadeln |
51 |
|
Fichtennadeln |
48 |
|
Fichtentriebe |
49 |
|
Fichtenrinde |
36 |
|
Pfirsich (Fruchtfleisch) |
40 |
|
|
|
|
Tierische Produkte |
|
|
Magermilchpulver |
42 |
|
Vollmilchpulver |
52 |
|
Rindfleisch, mager |
50 |
|
Rinderleber |
51 |
|
Fischfilet |
52 |
|
Human-Vollblut |
52 |
|
Schweine-Vollblut |
52 |
|
Austerngewebe |
46 |
|
Schweineniere |
49 |
|
Muschelgewebe |
41 |
|
Voll-Hühnerei |
50 |
|
|
|
|
Reine Kohlenhydrate |
|
|
Glucose-monohydrat |
37 |
|
Rohrzucker (Saccharose) |
42 |
|
Milchzucker (Lactose) |
42 |
|
Cellulose |
43 |
|
Stärke |
41 |
|
|
|
|
Fette |
|
|
Butter, pflanzliche Öle, |
|
|
pflanzliche Fette, Talg |
74...78 |
Auch wenn organische Matrixbestandteile bzw. deren Reaktionsprodukte mit HNO3 vollständig in Lösung überführt werden, so entstehen doch aus eiweißhaltigen Materialien und einigen Fetten lösliche organische Rückstände, die bei 180 oC von Salpetersäure nicht angegriffen werden[149] [150]:
- mehrfach ungesättigte Fettsäuren (Linol- und Linolensäure) werden durch HNO3 zu cis-1,2-Cyclopropandicarbonsäuren umgesetzt, die bei 180 oC gegen HNO3 beständig sind und somit im Aufschlußrückstand verbleiben.
- bestimmte Aminosäuren:
- Phenylalanin, dessen stabile Endprodukte diverse elektrochemisch aktive Nitrobenzoesäuren sind
- Histidin, dessen stabiles Endprodukt eine aromatische Carbonsäure ist.
- Tryptophan, dessen stabile Endprodukte mehrere aromatische Verbindungen, u. a. Carbonsäuren sind
- Methionin, das nur bis zur stabilen Methansulfonsäure umgesetzt wird.
Diese Abbauprodukte stören beispielsweise die inversvoltammetrische Bestimmung von Zn, Cd, Pb und Cu durch intensive Störsignale, erhöhen ebenfalls den Wasserstoffstrom und verschlechtern dadurch z.B. bei Zn das Signal/Untergrund-Verhältnis [151].
Bei Aufschlußzeiten von 10 – 12 Stunden bei 170 oC werden Aufschlußlösungen erhalten, die problemlos für AAS- oder ICP-AES-Messungen eingesetzt werden können, für voltammetrische Messungen aber selbst nach dem Abdampfen der Salpetersäure unbrauchbar sind, da die organische Matrix nicht vollständig zerstört wird [152].
Um dennoch zu brauchbaren Aufschlußlösungen mit diesen Apparaturen zu gelangen, wurden Verfahren mit verschiedenen Säuregemischen entwickelt, bei denen eine Nachbehandlung mit Perchlorsäure jedoch unbedingt erforderlich ist[152] [153]. Zur Nachbehandlung der Aufschlußlösung wird diese in einen Quarztiegel überführt und bei 180-200 oC zur Trockne eingedampft. Der Rückstand wird mit 0,5 ml Perchlorsäure mehrmals eingedampft und zum Schluß mit wenig Perchlorsäure und deionisiertem Wasser aufgenommen.
Jedoch wurden folgende Nachteile dieser Methode eingeräumt[152]:
- hoher Verbrauch an Säure (4 ml HNO3 und >2 ml HClO4)
- große Zahl an Einzelschritten
- Erhöhung der Blindwerte durch diese Prozeduren
Die Vorteile dieser Methode sind der geringe apparative Aufwand und relativ geringe Kosten.
In dieser Arbeit wurde für die Nachbehandlung der nach Druckaufschluß erhaltenen Probelösungen ein UV-Aufschluß durchgeführt.
Für die Durchführung des Druckaufschlusses wurden Autoklavensysteme mit PFA-Einsätzen (Perfluoralkoxy; Fa. CAT, Gummersbach) und PTFE-Einsätzen (Fa. PERKIN-ELMER, Überlingen) verwendet.
Diese konnten auf die maximal zulässige Betriebstemperatur von 160oC mittels eines zeitgesteuerten Laborofens in einem Abzugs erhitzt werden.
Für die Reinigung der PTFE-Einsätze zwischen zwei Aufschlußserien wurde mit 3 ml subboiled HNO3 8 h auf 160oC erhitzt.
Anschließend wurden die Gefäße gründlich mit Reinstwasser gespült und das Gesamtverfahren wiederholt.
Den biologischen Proben mit einer Einwaage von bis zu 300 mg wurden 3 ml subboiled HNO3 zugesetzt und zur Vorreaktion mehrere Stunden (meist über Nacht) stehengelassen. Bei Proben mit silikatischen Matrixanteilen wurden zusätzlich 100 µl HF als Aufschlußsäure zugesetzt [169].
Entstandene nitrose Gase wurden vor Einsetzen der Aufschlußgefäße in die Autoklaven unter dem Abzug der Cleanbench abgesaugt.
Anschließend wurde stufenweise erhitzt und bei 160oC für 10 Stunden aufgeschlossen (Tab. 34).
Tab.34 Durchführung des kombinierten Druck-UV-Aufschlusses
|
Arbeitsschritt |
Zeit (h) |
Addition |
|
Einwaage |
|
max.
300 mg Probe |
|
Zugabe Aufschlußsäure |
|
+3
ml subboiled HNO3, |
|
Vorreaktion |
> 8 |
|
|
Druckaufschluß |
10 |
160oC |
|
Umfüllen in UV-Röhrchen |
|
ad.
Vol. 10 ml |
|
UV-Bestrahlung |
4 |
+3 x 100µl H2O2 |
|
|
|
+100
µl HAS |
|
Umfüllen |
|
ad. Vol. 20 ml |
Die Druckaufschlüsse biologischer Proben führten zu gelblich verfärbten Lösungen, was auf den erwarteten unvollständigen Aufschluß der organischen Matrix und auf das Vorliegen von Nitrierungsprodukten hindeutet.
Die Aufschlußlösungen wurden daher in die UV-Probenröhrchen überführt, auf ein Volumen von 10 ml mit Reinstwasser aufgefüllt und nach Addition von 100µl H2O2 suprapur einer UV-Bestrahlung unterworfen.
Die Addition von 100µl H2O2 suprapur wurde während der UV-Bestrahlung im Abstand von einer Stunde für jedes Probengefäß wiederholt.
Der UV-Aufschluß wurde nach 4 Stunden mit der Zugabe von 100 µl Hydroxylammoniumsulfatlösung ((HONH3)2SO4 = 100 g/l) in die noch heiße Lösung beendet.
Mit den in Tab. 34 aufgeführten Arbeitschritten wurden farblose klare Lösungen erhalten, die meist ohne nennenswerte Störungen inversvoltammetrisch vermessen werden konnten.
Auch wenn für die Vorreaktion oder bei der Veraschung einschließlich Aufheiz- und Abkühlphase Nachtzeiten genutzt wurden, wird aus Tab. 34 ersichtlich, daß frühestens am Ende des auf die Probeneinwaage folgenden Tages die aufgeschlossenen Lösungen bereitgestellt werden konnten.
Zum vollständigen Umsatz biologischer Probenmaterialien mit Salpetersäure ist eine Temperatur von mindestens 300 oC erforderlich. Die resultierenden Aufschlußlösungen sind praktisch kohlenstofffrei; mehr als 99,9% des ursprünglich vorhandenen Kohlenstoffs der Probe sind nach 2-stündigem Aufschluß vom Analyten abgetrennt [151].
Aufschlüsse bei 300 oC können nur in Quarz- bzw. Suprasilgläsern, die mit Deckeln entsprechenden Materials verschlossen sind, durchgeführt werden.
Zwischen Gefäß und Deckel wird eine dünne Teflonfolie gelegt. Damit die Gefäße sowohl dem ansteigenden Innendruck standhalten als auch das Abdichten durch die Deckel gewährleistet bleibt, werden bis zu sechs Aufschlußgläser in einem beheizbaren Autoklaven einem Außendruck von 100 bar (N2, techn.) ausgesetzt. Die Steuerung des Zeit-Temperaturverlaufs und die Registrierung des Druckverlaufs kann durch eine Mikroprozessoreinheit gewährleistet werden. Die maximale Veraschungstemperatur beträgt 320 oC [148].
Biologisches Material verschiedener Matrices wurde von WÜRFELS et al [151] [154] [155] [156]nur mit 2 ml 65%iger Salpetersäure bei 320 oC Endtemperatur im 30-ml-Quarzgefäß aufgeschlossen. Allerdings müssen die Einwaagen biologischer Materialien einem reinen Kohlenstoffanteil von 100 mg C entsprechen, damit ein vollständiger HPA-Aufschluß (High Pressure Ashing, Fa. Kürner/Rosenheim) gesichert ist.
Mit dem Zusatz von 250 µl HClO4 und 50 µl H2SO4 bei einer Einwaage bis zu 400 mg Trockensubstanz wird die organische Matrix nach HASSE [152] aber immer so weit zerstört, daß die Aufschlußlösung für elektrochemische Bestimmungsmethoden brauchbar ist. Jedoch muß die Aufschlußlösung vor der eigentlichen Metallbestimmung auch bei dem HPA-Aufschluß zur Trockne eingeengt werden, um die gelösten Stickstoffoxide und eventuell noch vorhandene flüchtige organische Verbindungen zu entfernen [152] [149]. Durch Zugabe der Schwefelsäure fallen die Rückstände nach dem Abdampfen der HNO3 kristalliner aus und lösen sich leichter in verdünnter HClO4 oder HCl.
Obwohl die Reproduzierbarkeit der Meßergebnisse besser wird, besteht bei höheren Pb-Konzentrationen die Gefahr, daß Bleisulfat ausfällt und ein zu niedriger Bleiwert bestimmt wird [152].
Das Aufschlußverfahren wurde an einer Vielzahl von biologischen Materialien angewendet und neben Zn, Cd, Pb und Cu können auch Ni und Co adsorptionsvoltammetrisch gemessen werden [151] [152] [157]
Die Gesamtaufschlußzeit einschließlich Abkühlphase beträgt ca. 4 Stunden[152].
Die Kosten für den HPA sind aber mit. 60.000 – 70.000 DM bzw. für den Hochtemperatur-Verascher HTA mit ca. 50.000 DM [158] (Fa. Kürner Analysentechnik, Rosenheim) gegenüber einer Druckaufschlußapparatur (z.B. Fa. Berghof, Eningen) mit. 20.000 – 30.000 DM bei gleicher Probenanzahl wesentlich höher.
Die erste Arbeit zur Mikrowellenveraschung wurde 1975 von Koirtyohann et al publiziert [159], jedoch erst Ende der 80er Jahre von mehreren Arbeitsgruppen in den USA, Japan und Europa wieder aufgegriffen.
Die kommerziellen Mikrowellenöfen arbeiten mit einer Frequenz von 2,45 GHz und werden u.a. von den Firmen BERGHOF (Eningen), MLS (Fa. Milestone), CAL (Gummersbach), CEM (Moers), KÜRNER (Rosenheim), SPECTROTEC (Trebur) angeboten[160].
Aufgrund ihrer niedrigen Energie sind Mikrowellen nicht in der Lage, Valenzbindungen direkt aufzubrechen, da sie keine Schwingungs- oder Elektronenanregungen bewirken können, sondern lediglich Rotationsanregungen von Dipolen und Molekularbewegungen durch Wanderung von Ionen [161] [162]. Die Mikrowellenenergie wird durch Ionenleitung und Dipolrotation direkt in der Probelösung in Wärmeenergie umgewandelt.
Durch Druckaufschlußsysteme mit Mikrowellenanregung können unter Druckbedingungen die Aufschlußzeiten gegenüber den herkömmlichen Druckaufschlüssen wesentlich verringert werden. Durch die Verwendung von Einsätzen (Autoklaven) sind Hochdrucksysteme realisierbar, die einem hohen Druck standhalten und über die nötigen Sicherheitsmaßnahmen verfügen (10 bis zu 110 bar). Es lassen sich mit dieser Technik bereits komplexe geologische und biologische Matrices veraschen, wenn weniger anspruchsvolle Bestimmungsmethoden eingesetzt werden (F-AAS, GF-AAS, ICP-AES, ICP-MS) [163] [164]. Auch für die Analytik von Quecksilber ist der Einsatz von Mikrowellen-Druckaufschlußgefäßen erfolgreich durchführbar[165]
Die experimentellen Bedingungen für die Mikrowellendruckaufschlüsse sind je nach der zu veraschenden Matrix zu wählen [166] [167] [168], wobei fettreiche Proben beim Aufschluß aufgrund ihrer resistenten Inhaltsstoffe besondere Probleme bereiten.
Besonders elektrochemische Bestimmungen können nach Mikrowellenaufschluß durch Restkohlenstoff resistenter Verbindungen gestört werden. Da der Restkohlenstoffgehalt abhängig von der Veraschungstemperatur ist, schneiden nach DUNEMANN et al [163] Hochdrucksysteme besser ab als Niederdrucksysteme. Mikrowellensysteme hingegen schneiden etwas schlechter als die vergleichbaren herkömmlichen (konvektiven) Aufschlußtechniken ab.
Obwohl einige kommerziell erhältliche Mikrowellenaufschlußgeräte bis zu 10 Proben simultan aufschließen können, reicht die Mikrowellenenergie dieser Geräte im allgemeinen nicht, um die organische Matrix ausreichend zu zerstören [169] .
HASSE et al [169] beschreibt den Einsatz eines Druckaufschlußsystems mit druckgesteuerter (bis 85 bar) Mikrowellenheizung und Quarzeinsatz PMD (Pressurized Microwave Digestion, Fa. Kürner/Rosenheim) für eine Reihe von biologischen Materialien. Die “Qualität“ der Aufschlußlösungen wurde durch die Bestimmung von Pb, Cd, Cu, Ni und Co mittels DPASV und für As mittels Hydrid-AAS überprüft. Das nötige Aufschlußpotential ist demnach abhängig vom Typ der organischen Matrix.
Der Einsatz von Säurekombinationen (HNO3 / HClO4 / H2SO4) ist generell nötig, um einen hinreichend vollständigen Aufschluß der Probe zu erhalten. In einigen Fällen reichte die Mikrowellenenergie jedoch auch hier nicht aus, um mehr als eine Probe (bei max. zwei möglichen) Zeitgleich aufzuschließen [169].
Um die Mikrowellenenergie bei gleichzeitiger Druck- und Temperaturkontrolle gezielt auf die Reaktionslösung zu lenken, gehen die Entwicklungen weiter, indem z.B. Hochdruck-Teflonbomben und kombinierte Metallautoklaven mit integrierter Mikrowellenquelle entwickelt werden [170]. Durch einen Wellenleiter wird die Mikrowellenenergie direkt vom Mikrowellengenerator auf die Probe fokussiert. Die Temperatur des Autoklaven kann durch ein Kühlsystem während des Aufschlusses reguliert und anschließend vor dem Öffnen schnell auf Raumtemperatur gekühlt werden, bzw. der Innendruck wird wieder auf ein sicheres Niveau reduziert. Zudem ist durch die Verwendung moderner Polymere (Tetrafluoromethoxil: TFM-PTFE, Hoechst AG, Frankfurt) eine Aufschlußtemperatur bis 350 oC möglich.
Ein Vergleich des Restkohlenstoffgehaltes nach Aufschluß von Rinderleber (NIST-SRM 1577a) mit Salpetersäure wurde von MATUSIEWICS [170] mit der Hochdruckveraschung nach Knapp (HPA, s. Kapitel 12.3) durchgeführt. Die Oxidationseffizienz der fokussierten mikrowellenangeregten Aufschlußapparatur ist für diese Matrix nahezu mit der der HPA vergleichbar (99,4%, HPA: 99,6%), wird aber mit einem Zeitbedarf von nur etwa 3% gegenüber der thermischen Hochdrucktechnik (4 min, HPA: 120 min) angegeben [170].
Mit dieser “Pilotanlage“ konnte jedoch jeweils nur ein Aufschluß durchgeführt werden. Die angegebene Probenvorbereitungszeit von 10–12 min., einschließlich Abkühlzeit und Zubereitung der Meßlösung ist jedoch gegenüber den herkömmlichen Aufschlußmethoden bei nahezu vergleichbarer “Aufschlußqualität“ extrem verkürzt.
In der vorliegenden Arbeit wurde das kommerzielle Mikrowellendruckaufschluß-System MLS 1200 mega (Fa. Milestone) eingesetzt, mit dem bis zu 6 Proben simultan aufzuschließen sind.
Um für die inverse Voltammetrie brauchbare Aufschlüsse zu erhalten, müssen die gelösten Stickstoffoxide und eventuell noch vorhandene flüchtige organische Verbindungen nach Aufschluß entfernt werden (s. Kapitel 12.2). Zudem kann durch das Eindampfen der Aufschlußlösungen der Säuregehalt der meßfertigen Probenlösung exakt eingestellt werden, wodurch auf die Zugabe eines Puffers oder eine Verdünnung verzichtet werden kann.
Um das Umfüllen der Aufschlußlösungen und damit mögliche Blindwerte zu vermeiden, liegt es nahe, die Aufschlußlösungen nach Mikrowellendruckaufschluß auch in der Mikrowelle einzuengen.
Für das Einengen der nach dem Druckaufschluß erhaltenen Lösungen stand ein Eindampfrotor MCR-6E (Fa. Milestone) zur Verfügung.
Die Aufschlußgefäße werden zum Eindampfen nach Beendigung des Druckaufschlusses ohne Deckel und mit einem aus dem Material Weflon® gefertigten Mantel in den Eindampfrotor gestellt. Die Weflon-Mäntel sind aus PTFE mit eingelagertem Kohlenstoff gefertigt und erwärmen sich in der Mikrowelle (Abb.45).
Um während des Mikrowellenbetriebes die entstehenden Säuredämpfe aus den Aufschlußgefäßen zu entfernen und eine Durchmischung des Dampfraumes zu erreichen, wird der Eindampfrotor mit dem Gaswäscher B-412 (Fa. Büchi) als Absaugeinheit betrieben.
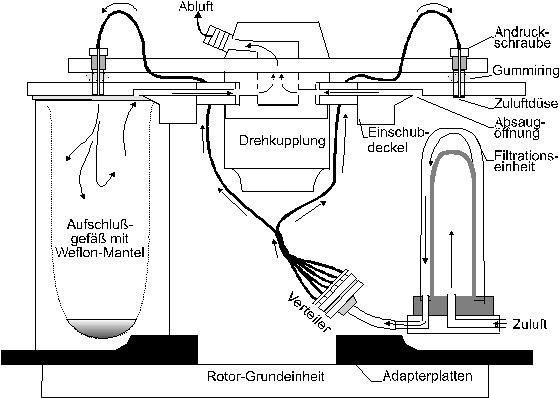 |
Abb.45 Schematische Darstellung des Eindampfrotors MCR 6E |
Die unzureichende Luftführung führte jedoch zunächst zu einem erschwerten Abdampfen und zu einer Kontamination einzelner Proben. Da der Gefäßrand die Absaugöffnungen im Deckel des Abdampfrotors verdeckte, wurden die Adapterplatten gelockert und weiter in den Innenbereich des Rotors verschoben.
Die in der Höhe geringfügig flexiblen Einschubdeckel wurden durch Gummiringe und Andruckschrauben auf die Aufschlußgefäße angepreßt, um das Ansaugen von Nebenluft, wodurch neben einer möglichen Kontamination die Luftführung unterbrochen wird, zu verhindern.
Zudem wurde die angesaugte Zuluft über eine Filtrationseinheit aufgereinigt, bevor sie über den Verteiler zu den Zuluftdüsen der Einschubdeckel gelangte. Hierzu wurde die Filtrationseinheit AGF-PF (Fa. Bühler) aus PVDF (Polyvinylidenfluorid) für den Mikrowellenbetrieb aus PTFE nachgebaut, da die Filtrationseinheit konstruktionsbedingt im Inneren des Eindampfrotors betrieben werden mußte. Als Filtereinsatz wurde ein S2-Glasfaserfilter mit 2µm Porenweite (Fa. Bühler) eingesetzt.
Für die Durchführung der Probenvorbereitung wurde das Mikrowellendruckaufschluß-System MLS 1200 mega (Fa. Milestone) mit einer Mikrowellenfrequenz von 2,45 GHz eingesetzt. Das System wird mit einer Absaugeinheit EM-30 (Fa. Milestone) betrieben.
Die 100 ml Aufschlußgefäße aus TFM-PTFE sind für Druckverhältnisse bis zu 110 bar ausgelegt. Mit dem HRP-1000/6-Rotor ist die gleichzeitige Durchführung von sechs Aufschlüssen möglich.
Die eingestrahlte Leistung kann für die Erstellung von Aufschlußprogrammen zwischen 250 Watt bis 1000 Watt in 10 Watt-Inkrementen mit der entsprechenden Bestrahlungsdauer variiert und abgespeichert werden. Maximal sind 10 Programmschritte möglich.
Die Energie wird bei der 250 Watt-Stufe ungepulst eingestrahlt, während bei Leistungen über 250 Watt 1000 Watt gepulst werden. Als Abkühlphase dienen Null-Watt-Schritte, um die Sicherheit zu erhöhen.
Für die Reinigung der PTFE-Einsätze zwischen zwei Aufschlußserien wurde mit 2,5 ml subboiled HNO3 ein Reinigungsprogramm (Tab. 35) durchgeführt:
Tab 35 Reinigungsaufschluß für Mikrowelleneinsätze |
|
Programmschritt |
Zeit [min] |
Leistung [W] |
|
1 |
10 |
250 |
|
2 |
1 |
0 |
|
3 |
5 |
500 |
|
4 |
1 |
0 |
|
5 |
5 |
700 |
Anschließend wurden die Gefäße gründlich mit Reinstwasser gespült und das Gesamtverfahren zweimal wiederholt.
Druckaufschlüsse biologischer Proben, die zunächst nur mit Salpetersäure durchgeführt wurden, führten zu gelblich bis grünlich verfärbten Lösungen. Dies deutet auf einen unvollständigen Aufschluß der organischen Matrix und auf das Vorliegen von Nitrierungsprodukten hin, wodurch die voltammetrische Bestimmung, wie in Kapitel 12 beschrieben, erschwert oder sogar unmöglich wird.
Den biologischen Proben mit einer Einwaage von ca. 200 mg wurden 3 ml subboiled HNO3 und 250 µl HClO4 zugesetzt und zur Vorreaktion mehrere Stunden (meist über Nacht) stehengelassen. Proben mit silikatischen Matrixanteilen, wie beispielsweise die Standardreferenzmaterialien GBW0805 Tee und GBW08504 Kohl, wurden zusätzlich 100 µl HF als Aufschlußsäure zugesetzt [169]. Während der Vorreaktion waren die Aufschlußgefäße nur durch die aufliegenden Deckel verschlossen. Entstandene nitrose Gase wurden vor Einsetzen der Aufschlußgefäße in den Rotor unter dem Abzug der Cleanbench abgesaugt und die Proben mit folgendem Mikrowellenprogramm (Tab. 36) einem Aufschluß unterzogen [171]:
Tab.36 Mikrowellenprogramm für biologische Proben |
|
Programmschritt |
Zeit [min] |
Leistung [W] |
|
1 |
1 |
250 |
|
2 |
3 |
0 |
|
3 |
3 |
250 |
|
4 |
3 |
0 |
|
5 |
3 |
500 |
|
6 |
3 |
0 |
|
7 |
3 |
800 |
|
8 |
3 |
0 |
|
9 |
3 |
1000 |
|
10 |
10 |
0 |
Es erfolgte ein Abkühlen des Aufschlußrotors, was durch Plazieren des Rotors in ein Kaltwasserbad beschleunigt werden konnte (~ 30 min.), bevor die unter Druck stehenden Gefäße ohne Abblasen und somit ohne Verlust an Aufschlußlösung geöffnet werden konnten. Nitrose Gase wurden evtl. erneut abgesaugt und das Aufschlußverfahren fortgesetzt (Tab. 37)
Tab. 37 Fortsetzung des Aufschlußprogramms |
|
Programmschritt |
Zeit [min] |
Leistung [W] |
|
11 |
3 |
1000 |
|
12 |
3 |
0 |
|
13 |
5 |
1000 |
|
14 |
3 |
0 |
|
15 |
5 |
1000 |
Die Kombination von Aufschlußsäuren und/oder die Zugabe von 250 µl Wasserstoffperoxid und einer Fortsetzung des Aufschlußprogramms (Tab. 37) führte zwar schon zu klaren, aber immer noch unvollständigen Aufschlüssen und in der Folge zu nicht auswertbaren Voltammogrammen.
Die Aufschlußgefäße wurden daher nach dem Druckaufschluß ohne Deckel mit einem aus Weflon® gefertigten Mantel versehen in den Eindampfrotor gestellt. Das Eindampfprogramm (Tab. 38) mußte auf das aktuelle Volumen der Lösung in den Aufschlußgefäßen abgestimmt sein, um den Verlust von Elementspuren zu vermeiden, die im Säuredampf enthalten sind, wenn die Lösung unter Druck zu Sieden beginnt.
Tab. 38 Eindampfprogramm für biologische Proben |
|
Programmschritt |
Zeit [min] |
Leistung [W] |
|
14 |
1 |
500 |
|
15 |
17 |
250 |
|
16 |
5 |
100 |
Anschließend wurden die Innenwände der Aufschlußgefäße mit 5 ml Reinstwasser abgespült und mit dem Eindampfprogramm (Tab. 39) auf das Perchlorsäurevolumen von etwa 250 µl eingeengt.
Tab. 39 Fortsetzung des Eindampfprogramms |
|
Programmschritt |
Zeit [min] |
Leistung [W] |
|
17 |
2 |
250 |
|
18 |
10 |
100 |
In diesem letzten Schritt können die evtl. noch vorhandenen organischen Bestandteile durch die Perchlorsäure in konzentrierter Form aufoxidiert werden.
Die Aufschlußlösungen sind nach Beendigung des kombinierten Aufschluß- und Eindampfverfahrens farblos und klar und können in der Regel problemlos mit der Inversvoltammetrie gemessen und ausgewertet werden.
Die Aufschlußlösungen wurden in der Regel auf ein Endvolumen von 20 ml aufgefüllt und konnten bei einem pH-Wert von 1,6 – 1,8 unverdünnt voltammetrisch gemessen werden.
[130] Saur, D.: Aufschlußtechniken in der voltammetrischen Spurenanalyse, in: Probenvorbereitungstechniken in der voltammetrischen Spurenanalyse, Metrohm Monographien, Herisau (1991).
[131] Sichere Chemiearbeit 35, (1983) 85.
[132] Würfels, M.; Jackwerth, E.: Untersuchungen zur Kohlenstoffbilanz beim Aufschluß biologischer Probenmaterialien mit Salpetersäure, Fresenius Z. Anal. Chem. 322 (1985) 354-358.
[133] DIN 38406 Teil 16: Deutsches Einheitsverfahren zur Wasser-, Abwasser- und Schlammuntersuchung, Kationen (Gruppe E), Bestimmung von 7 Metallen (Zn, Cd, Cu, Tl, Ni, Co) mittels Voltammetrie (E16), Beuth Verlag, Berlin (1990).
[134] Mart, L.: Minimization of Accuracy Risks in Voltammetric Ultratrace Determination of Heavy Metals in Natural Waters. Talanta 29 (1982), pp. 1035 – 1040.
[135] Nürnberg, H.W.; Valenta, P.: Potentialities and Applications of Voltammetry in Chemical Speciation of Trace Metals in the Sea, in: C.S. Wong; E. Boyle; K.W. Bruland; J.D. Burton; E.D. Goldberg (Eds.): Trace Metals in Sea Water, pp. 671-679, Plenum Press, New York-London (1983).
[136] Nürnberg, H.W.; Raspor,B.: Applications of Voltammetry in Studies of the Speciation of Heavy Metals by Organic Chelators in Sea Water. Environm. Technol. Letters 2 (1981), pp. 337 - 346.
[137] Valenta, P.: Voltammetric Studies on Trace Metal Speciation in Natural Waters, Part I. Methods, pp. 49-69; Nürnberg, H.W.: Part II Applications and Conclusions for Chemical Oceanography and Chemical Limnology, pp. 211 - 230 in: G.G. Leppard (Ed.): Trace Element Speciation in Surface Waters and ist Ecological Implications, Plenum Publ. Corp., New York - London (1983).
[138] Mart, L.; Nürnberg, H.W.; Valenta, P.: Voltammetric Ultratrace Analysis with a Multicell System designed for Clean Bench Working; Fresenius Z. Anal. Chem. 300, 350-362, (1980).
[139] Sipos, L.; Golimowski, J.; Valenta, P.; Nürnberg, H.W.: New Voltammetric Procedure for The Simultaneous Determination of Copper and Mercury in Environmental Samples : Freseniuz Z. Anal. Chem.298 (1979) 1-8.
[140] Mart, L.: Minimization of Accuracy Risks in Voltammetric Ultratrace Determination of Heavy Metals in Natural Waters. Talanta 29 (1982) 1035-1040.
[141] Pilhar, B.; Valenta, P.; Nürnberg, H.W.: New High-Performance Analytical Procedure for the Voltammetric Determination of Nickel in Routine Analysis of Waters, Biological Materials and Food: Fresenius Z. Anal. Chem. 307, 337 – 351 (1981)
[142] Florence, T.M.:Development of Physiko-Chemical Speciation Procedures to Investigate the Toxicity of Copper, Lead, Cadmium and Zink towards Aquatic Biota, Analytica Chimica Acta, 141 (1982) 73 - 94
[143] Kolb, M.; Rach, P.; Schäfer, J.; Wild,A.: Investigations of oxidative UV photolysis, I. Sample preparation for the voltammetric determination of Zn, Cd, Pb, Cu, Ni, and Co in Waters, Fresenius J. Anal. Chem. 342 (1992) 341-349
[144] Tauchlampen für die Photochemie, Technisches Datenblatt für Strahlenquellen, Heraeus Nobellight GmbH, D 87520 Kleinostheim, Oktober 1992.
[145] Aufschluß biologische Matrices für die Bestimmung sehr niedriger Spurenelementgehalte bei begrenzter Einwaage mit Salpetersäure unter Druck in einem Teflongefäß, Fresenius Z. Anal. Chem. 260 (1972) 207-209
[146] Schrammel, P.; Wolf, A.; Seif, R.; Klose, B.J.: Eine neue Apparatur zur Druckveraschung von biologischem Material, Fresenius Z. Anal. Chem. 302 (1980) 62
[147] Knapp, G.: Methoden zum Aufschluß organischer Materialien für die Elementspurenanalyse, in: B.Welz, Atomspektrometrische Spurenanalytik, Verlag Chemie, Weinheim/Bergstraße (1982)
[148] Knapp, G.: Der Weg zu leistungsfähigen Methoden der Elementspuren-analyse in Umweltproben: Fresenius Z. Anal. Chem. 317 (1984) 213
[149] Würfels, M.: Dissertation, Ruhr Universität, Bochum, 1988, in: Würfels, M.: Säureaufschluß biologischer Proben zur Elementbestimmung, Kürner Analysentechnik, LABO 3 (1989) 1-7
[150] Würfels, M.; Jackwerth, E.; Stoeppler, M.; Fresenius J Anal Chem, 330, 159 (1988), in: Würfels, M.: Säureaufschluß biologischer Proben zur Elementbestimmung, Kürner Analysentechnik, LABO 3 (1989) 1-7
[151] Würfels, M.; Jackwerth, E.; Stoeppler, M.: Probenvorbehandlungsstudien mit biologischen und Umweltmaterialien; V. Zum Problem der Störung inversvoltammetrischer Spurenanalysen nach Druckaufschluß biologischer Probenmaterialien. Fresenius Z. Anal. Chem. 329 (1987) 459,
[152] Hasse, S.: Voltammetrische Schwermetallbestimmungen in Biomatrices nach Druckaufschlüssen mit Nachbehandlung, in: Probenvorbereitungs-techniken in der voltammetrischen Spurenanalyse, Metrohm Monographien, Herisau (1991).
[153] Hasse, S.; Schrammel, P.: Voltammetric Determination of Cd, Cu, Co, Ni and Pb in Milkpowder and other biological materials with Special Regard to the Ashing Method, Mikrochimica Acta (Wien) III (1983) 449-455.
[154] Würfels, M.; Jackwerth, E.; Stoeppler, M.: Residues from biological Materials after Pressure Decomposition with Nitric Acid. Part 1: Carbon Conversion during Sample Decomposition Anal. Chim. Acta 226 (1989) 1-6.
[155] Würfels, M.; Jackwerth, E.; Stoeppler, M.: Residues from biological Materials after Pressure Decomposition with Nitric Acid. Part 2: Identifikation of the Reaction Products Anal. Chim. Acta 226 (1989) 17-30
[156] Würfels, M.; Jackwerth, E.; Stoeppler, M.: Residues from biological Materials after Pressure Decomposition with Nitric Acid. Part 3: Influence of Reaction Products on Inverse Voltammetric Element Determination Anal. Chim. Acta 226 (1989) 31-41.
[157] Schrammel, P.; Hasse, S.; Knapp, G.: Einsatz des Hochdruckveraschers HPA nach Knapp für die voltammetrische Bestimmung von Spurenelementen in biologischem Material, Fresenius Z. Anal. Chem 326 (1987) 142-145.
[158] Kürner, H.: Druckaufschluß in der Voltammetrie – Probleme und Lösungen in: Metrohm Monographien, Herisau (1991).
[159] Abu-Samra, A.; Morris, J.S.; Koirtyohann, S.R.: Wet ashing of some biological samples in a microwave oven. Anal. Chem. 47 (1975) 1475.
[160] Dunemann, L.: Marktübersicht: Aufschlußmethoden für die Spurenanalytik, Nachr. Chem. Tech. Lab. 39 (1991) M1.
[161] Neas, E.D.; Collins, M.J.: Microwave heating – Theoretical concepts and equipment design. In: Introduction to Microwave Sample Preparation, Kingston, H.M.; Jassie L.B. (eds.), American Chemical Society, Waschington DC, 7 (1988).
[162] Nadkarni, R.A.: Applications of microwave oven Sample dissolution in analysis. Anal. Chem. 56 (1984) 2233.
[163] Dunnemann, L.: Neue mikrowellenunterstützte Aufschlußmethoden zur Spurenanalytik in biologischen Matrices. GIT-Fachz. Lab., 10 (1993) 854 – 858.
[164] Lippo, H.; Särkälä, A.: Microwave Dissolution Method for the Determination of Heavy Metals in Biomonitors Using GFAAS and Flame AAS; J. Anal. At. Spectrosc. 8 (1995) 154-157.
[165] Navarro, M.; Lopez, M.C.; Lopez, H.; Sanchez, M.: Microwave dissolution for the determination of mercury in fish by cold vapour atomic absorption spectrometry, Anal. Chim. Acta 257 (1992) 155.
[166] Dunnemann, L.; Meinerling, M.: comparison of different microwave-based digestion techniques in view of their application to fat-rich foods, Fresenius J. Anal. Chem. 342 (1992) 714-718.
[167]> Zunk, B.: Mikrowellenaufschluß zur Bestimmung von Spurenelementen in Pflanzenmaterial, Anal. Chim. Acta. 236 (1990) 337-349.
[168] Xu, L.Q.; Shen, W.X.: Rapid decomposition method for silicate Samples, Fresenius Z. Anal. Chem. 333 (1989) 108.
[169] Schrammel, P.; Hasse, S.: Destruktion of organic materials by pressurized microwave digestion, Fresenius J. Anal. Chem. 346 (1993) 794 – 799.
[170] Matusiewicz, H.: Development of a High Pressure/Temperature Focused Microwave Heated Teflon Bomb for Sample Preparation, Anal. Chem. 66 (1994) 751 – 755.
[171] Agger, J.; Bredthauer, U.; Wieberneit, N.; Dannecker, W.: Applicabillity of a fully automated voltammetric analysis system for the determination of trace and ultratrace amounts of heavy-metals in biological and environmental samples. In: Proceedings of the International Conference "heavy metals in the environment" in Hamburg 18.9-22.9.1995. Hrsg.: R.D. Wilken, U.Förstner, A.Knöchel, Vol.2 (1995) 357-360.