Taucht eine Metallelektrode in eine Elektrolytlösung, so werden durch die Ladung auf dem Metall Ionen entgegengesetzter Ladung an die Grenzfläche angezogen.
Im idealen Fall werden bei hinreichender Spannung die an die tropfende Quecksilberelektrode gelangenden Depolarisatorteilchen (elektroaktive Species) M+ auf der Grundlage der Faradayschen Gesetze sogleich entsprechend M+ + e- ® Mo elektrolysiert.
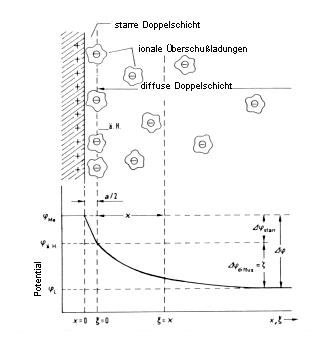
Das Potential in der Lösung verändert sich in unmittelbarer Nähe der Elektrode (Abb. 2). Direkt an der Elektrodenoberfläche werden Ionen mit der Elektrodenladung entgegengesetzter Ladung spezifisch adsorbiert. Es entsteht die sogenannte starre Doppelschicht. Die durch die Ladungsschwerpunkte der angelagerten Ionen verlaufende Ebene wird als äußere Helmholtz-Ebene bezeichnet. Hier fällt das Potential linear ab. Jenseits der starren Doppelschicht liegt in Elektodennähe ein weiterer Überschuß an Ionen, die der Elektode entgegengesetzt geladen sind, vor. Auf Grund der Ionen-Wärmebewegung tritt aber keine spezifische Adsorption auf, so daß diese Schicht als diffuse Doppelschicht bezeichnet wird. Hier erfährt das Potential einen exponentiellen Abfall [18].
 |
|
Abb.2: Potentialverlauf durch die
elektrolytische Doppelschicht. |
Diese Phasengrenze kann auch als Kondensator aufgefaßt werden, wobei sich die Doppelschichtkapazität mit dem Potential und der Elektrodenoberfläche ändert. Dadurch entsteht ein kapazitiver Ladestrom (Grundstrom), der dem Meßsignal (Faradayscher Strom) überlagert ist. Durch den Elektronentransfer an der Elektrode und der damit verbundenen Umsetzung der Teilchen M+ stellt sich ein Konzentrationsgefälle dieser Species zwischen der Elektroden-oberfläche und der Lösung ein.
Die Konvektion (durch Rühren) in der Lösung führt den Analyten an die Diffusionsschicht des Tropfens heran. Innerhalb der Diffusionsschicht, also in unmittelbarer Nähe der Tropfenoberfläche, erfolgt der Stofftransport dann ausschließlich durch Diffusion. Wird eine elektrochemisch aktive Substanz in der Nähe der Elektrode umgesetzt, so bestimmt der Diffusionsvorgang, d.h. die Diffusionsgeschwindigkeit, den Stromfluß. Der Diffusionsgrenzstrom wird bei genügendem Überschreiten des Halbstufenpotentials erreicht.
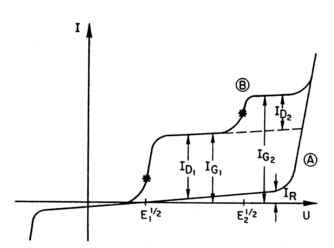 |
Abb.3: Darstellung eines
Gleichstrompolarogramms aus RACH [19]
|
Die Konzentrationsabhängigkeit des Faradayschen Diffusionsstromes ist im allgemeinen die Grundlage der quantitativen polarographischen/ voltammetrischen Analyse. Die mathematischen Grundlagen für die Berechnung des Konzentrationsgradienten am wachsenden Quecksilbertropfen wurden von ILKOVIC hergeleitet.
Befindet sich in der Elektrolytlösung keine im untersuchten Spannungsbereich reduzierbare oder oxidierbare Substanz, so wird nur ein kleiner, mit negativerer Spannung schwach ansteigender Strom registriert. (s. Abb. 3A). Dieser Strom wird als Reststrom (IR) bezeichnet. Die Arbeitselektrode kann in diesem Fall als polarisiert betrachtet werden. Eine ideal polarisierte Elektrode nimmt gemäß Definition jede ihr aufgeprägte Spannung an, ohne daß hierbei ein Strom fließt.
Enthält die Elektrolytlösung jedoch eine im untersuchten Spannungsbereich reduzierbare oder oxidierbare Substanz, so wird beim Erreichen der Spannung, bei welcher die Substanz an der Elektrode reduziert oder oxidiert wird, ein Strom fließen. Dieser Strom wird bei weiterer Änderung der Spannung zunächst stark zunehmen, dann abflachen und schließlich einem Grenzwert zustreben, bei welchem alle an der Elektrodenoberfläche eintreffenden Substanzen sofort reduziert bzw. oxidiert werden (Abb. 3B).
Im Bereich des Stromanstiegs ist die Elektrode depolarisiert. Daher wird die den Strom verursachende Substanz "Depolarisator" genannt. Beim Grenzstrom gelangt die Substanz ausschließlich durch Diffusion an die Elektroden-oberfläche. Der Stofffluß wird durch die Fickschen Gesetze beschrieben und ist der Konzentration in der Lösung direkt proportional. Die Fickschen Gesetze sind die Grundlagen zur quantitativen Beschreibung einer linearen Diffusion.
Wird die Messung mit Hilfe einer Quecksilbertropfelektrode (Dropping Mercury Elektrode: DME) aufgenommen, so wird der Strom bei weiteren Änderungen der Spannung so lange bei diesem Grenzwert bleiben, bis die Zersetzungs-spannung (UZ) einer anderen elektroaktiven Substanz erreicht wird. Die Elektrode ist in Bezug auf die zuvor umgesetzte Substanz polarisiert (Abb. 4a).
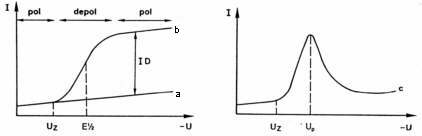 |
Abb.4 Verlauf der Strom-Spannungskurve an der
Quecksilbertropfelektrode (a und b) |
Da der Stoffluß durch den diffusionskontrollierten Strom (ID) aus der Elektrodenreaktion wiedergegeben wird, ist dieser Strom ein direktes Maß für die Konzentration der Substanz in der Lösung. Die Spannung, bei welcher der diffusionskontrollierte Grenzstrom zur Hälfte ausgebildet ist, wird als Halbstufen- bzw. Halbwellen-Potential (E˝) bezeichnet und dient zur qualitativen Charakterisierung des Analyten. Den depolarisierten Teil der Strom-Spannungs-Kurve bezeichnet man als polarographische Stufe oder Welle.
Wird die Messung mit Hilfe einer Fest-Elektrode vorgenommen, so wird sich die Diffusionsschicht gemäß dem 2. Fickschen Gesetz nach Erreichen des Grenzstroms mit der Zeit vergrößern, d. h. der Stoffluß und damit der diffusionskontrollierte Strom wird asymptotisch abnehmen. Es wird keine Stufe, sondern eine glockenförmige Kurve (Peak) erhalten, wobei der maximale Wert des diffusionskontrollierten Stroms (Ip) der Konzentration des Analyten direkt proportional und die Spannung beim Strompeak (Up) charakteristisch für die Substanz ist (Abb.4b).
[18] Hamann, C.H.; Vielstich, W.: Elektrochemie I, Verlag Chemie, Weinheim (1985).
[19] Rach, P.; Seiler, H.: Polarographie und Voltammetrie in der Spurenanalytik, Hüthig Verlag, Heidelberg (1985).